
PEER REVIEWED
Stargardt Disease
Raman Bhakhri OD, FAAO, Sarah Neidermann, and Gabriella Vivacqua
Abstract
Stargardt disease is an inherited retinal condition. With an incidence of 8-10 per 100,000 persons, it is the most common juvenile macular degeneration.1 The majority of cases present in an autosomal recessive fashion and are due to mutations in the ABCA4 gene. The condition tends to manifest at an early age; however, large amounts of heterogeneity are seen in terms of genotype and phenotype thus the condition can have a later onset with variable clinical findings.2 The purpose of this case report is to present two different phenotypic presentations of Stargardt disease that were diagnosed based on a combination of multimodal imaging and genetic testing. A comprehensive review of the etiology, clinical features, treatment and management of the disease is also presented.
Key Words: Stargardt, ABCA4, fundus autofluorescence, inherited retinal diseases, optical coherence tomography
Introduction
Stargardt disease is a genetic condition affecting the retina and is one of the most commonly inherited retinal diseases.3 The condition affects the macula and leads to the damage and eventual loss of photoreceptors and underlying retinal pigment epithelium (RPE), which causes decreased central vision and possible central/paracentral scotomas. Although age of onset can vary, Stargardt disease typically begins to manifest during early childhood. Onset during early or late adulthood is less common, but later onset is associated with a better prognosis.2,4,5
The disorder is an inherited retinal disease with the majority of cases inherited in an autosomal recessive pattern secondary to a genetic mutation(s) within the ABCA4 gene. The condition is referred to as Stargardt disease 1 (STGD1) if the ABCA4 gene is implicated. The ABCA4 gene encodes for the ABCA4 protein, which is responsible for removing toxic substances from the photoreceptors.6-8 In a patient with this mutation, the ABCA4 protein is defective, leading to accumulation of a toxic substance known as lipofuscin within the retinal cells. This ultimately results in cell death.9 Rarer autosomal dominant forms of inheritance also exist for Stargardt disease.
There is no treatment for STGD1 at this time. However, clinical trials are currently investigating gene replacement therapy, stem cell therapy and pharmacological interventions. This case report highlights two STGD1 patients with different phenotypic presentations. It also reviews pathophysiology, differential diagnosis, multimodal imaging results and potential treatment and rehabilitation options.
Case Description
Patient 1
A 21-year-old African American female presented for a comprehensive examination complaining of longstanding distance blur in both eyes. Her ocular history was remarkable for probable STGD1, which was diagnosed at an outside office 3 years prior. At that visit glasses were prescribed and no follow-up was scheduled. The patient’s medical history was unremarkable. Her family’s medical and ocular histories were unremarkable. She denied any medication use or any allergies. Best-corrected visual acuity was 20/400 in the right and left eye. The patient noted that her visual acuities were stable to the previous examination’s findings. Entrance testing and slit lamp examination were normal. Intraocular pressure measured 15 mmHg in the right and left eye. A dilated fundus exam revealed a cup-to-disc ratio of 0.4/0.4 horizontally and vertically in each eye. Both optic nerves were pink with distinct margins. Macular atrophy was present with surrounding pisciform flecks in both eyes as evidenced by fundus photographs (Figure 1). A spectral domain optical coherence tomography (SD-OCT) scan was performed and revealed RPE atrophy along with focal and hyper-reflective RPE thickening and disruption. Ellipsoid zone (EZ) atrophy with extension nasal and temporal to the fovea was evident in both eyes (Figure 2). Fundus autofluorescence (FAF) showed large amounts of central hypo-autofluorescence corresponding to the RPE atrophy. Hyper-autofluorescence of the pisciform flecks was also observed in both eyes with a surrounding hyper-autofluorescent border. Peripapillary sparing on FAF was also visible (Figure 3). Based on her early age at disease onset and fundus findings, the patient was diagnosed with STGD1. Unfortunately, genetic testing was not readily available at this time. The patient was referred for low vision rehabilitation (LVR) services but was lost to follow-up.
 Figure 1. Fundus photographs of the right (A) and left (B) eye of patient 1 demonstrating macular atrophy (yellow arrows) with surrounding pisciform fleck accumulation (blue arrows). |
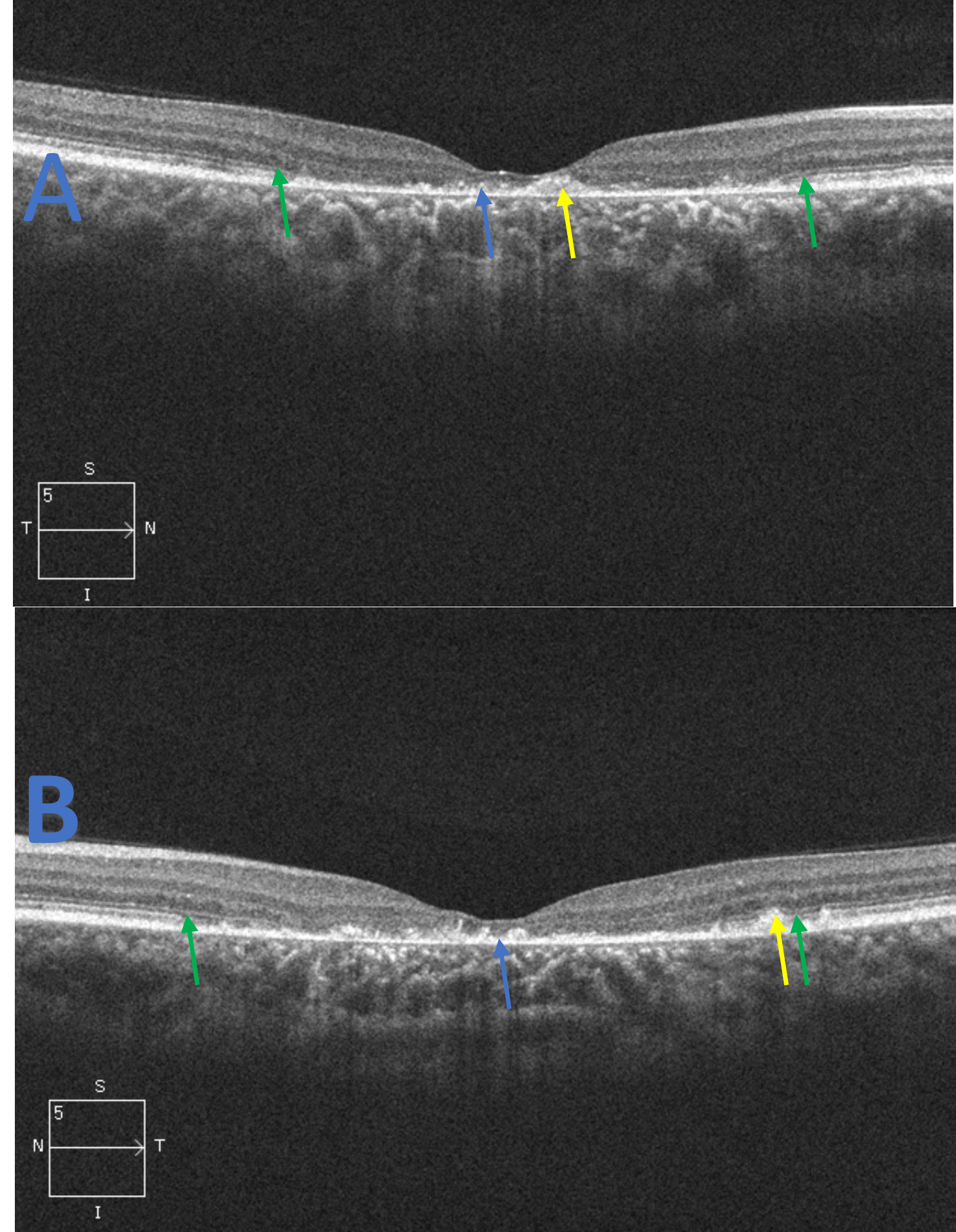 Figure 2. Spectral domain optical coherence tomography raster scans of the macula, right eye (A) and left eye (B), for patient 1. The scans reveal central RPE atrophy (blue arrow) along with focal and hyper-reflective RPE thickening and disruption (yellow arrow). Foveal ellipsoid zone (EZ) atrophy is evident with extension nasal and temporal to the fovea in both eyes. The junction of the preserved and damaged EZ is indicated (green arrow). These findings correspond to the appearance of the fundus in Figure 1 and the central hypo-autofluorescence observed in Figure 3. Click to enlarge |
 Figure 3. Fundus autofluorescence imaging of the right (A) and left (B) eye of patient 1 revealing hypo-autofluorescence at the macula (yellow arrows) corresponding to the macular/retinal pigment epithelium (RPE) atrophy observed in Figures 1 and 2. Scattered areas of hyper-autofluorescence (blue arrows), corresponding to RPE disruption and lipofuscin accumulation/pisciform flecks seen in Figure 1 and 2, are observed around the RPE atrophy. A hyper-autofluorescent border is also noted surrounding the central lesions (yellow circles) in addition to peripapillary sparing (blue circle). Click to enlarge |
Case Description
Patient 2
A 45-year-old African American female first presented with a chief complaint of longstanding blurry vision. Her medical history was unremarkable and she was not using medications. However, she reported a history of prior cigarette smoking (1/2 pack a day for 15 plus years). Her family’s medical and ocular histories were unremarkable. Best-corrected visual acuity was 20/50 and 20/30 in the right and left eye, respectively. Entrance testing and slit lamp examination were unremarkable. Intraocular pressure measured with Goldmann tonometry was 17 mmHg in each eye. The dilated fundus examination showed a cup-to-disc ratio of 0.3/0.3 horizontally and vertically in each eye. Both optic nerves were pink with distinct margins. Fundus photographs of the macula in each eye showed RPE atrophy with adjacent RPE hyperplasia. Pisciform-like deposits were noted extending into the arcades and midperiphery in both eyes (Figure 4). The periphery was unremarkable in both eyes. SD-OCT scans of both eyes showed larges areas of RPE atrophy nasal to the fovea with minimal foveal preservation. RPE and EZ disruption were also evident on the SD-OCT scans, involving and temporal to the fovea (Figure 5). The macular findings and the patient’s relatively good entering visual acuities were suggestive of an inherited retinal disease. A tentative diagnosis of fundus flavimaculatus (FF), a subtype of STGD1, was made. Unfortunately, genetic testing and FAF were not available in the clinic at that time. The patient declined a referral to a retina specialist as she had minimal complaints and relatively good visual acuity. She was therefore advised to return yearly to be monitored for disease progression.
The patient was lost to follow-up and returned 8 years later. Her medical and ocular histories were unchanged. Best-corrected visual acuity was 20/800 and 20/200 in the right and left eye, respectively. Entrance testing and slit lamp examination were unremarkable. Based on Goldmann tonometry, intraocular pressure was 16 mmHg in each eye. The dilated fundus examination showed a cup-to-disc ratio of 0.3/0.3 horizontally and vertically in each eye. Both optic nerves were pink with distinct margins. The macula in each eye showed extensive RPE atrophy, adjacent RPE hyperplasia and retinal flecks extending into the midperiphery as seen with ultra-widefield imaging (Figure 6). FAF showed large areas of central macular hypo-autofluorescence (Figure 7) corresponding to the macular atrophy observed in Figure 6. Mixed hyper- and hypo-autofluorescence was noted surrounding the central macular hypo-autofluorescence and likely represented active lipofuscin accumulation (hyper) and reabsorbed lipofuscin/RPE atrophy (hypo). Peripapillary sparing was also visible in both eyes. Generalized RPE involvement was indicated by global hyper-autofluorescence extending into the arcades. Progression of the atrophy was noted when compared to the fundus photos from her initial visit (Figure 8). Genetic testing confirmed STGD1, subtype FF, as the results revealed mutations for ABCA4 c.570+1798A>G (intron variant) and ABCA4 c.5114G>A (missense mutation). The patient was advised to schedule an appointment for genetic counseling based on her test results and was strongly advised to seek LVR services due to her vision loss. The patient declined LVR services and was asked to return in 1 year to be monitored for progression and to reconsider LVR.
 Figure 4. Fundus photos upon initial presentation of patient 2. Retinal pigment atrophy (yellow arrows) is evident in the right eye (A) and left eye (B) with partial foveal preservation in both eyes. Large amounts of pisciform fleck are observed extending into the arcades of both eyes (blue arrows). Click to enlarge |
 Figure 5. Spectral domain optical coherence tomography raster scans of the macula from the right eye (A) and left eye (B) of patient 2. Scanning reveals retinal pigment epithelium atrophy, more so nasal to the fovea in both eyes (blue arrow), which corresponds to the fundus appearance in Figure 4. Retinal pigment disruption is noted as hyper-reflectivity (yellow arrow). Ellipsoid zone compromise, but not complete atrophy, is noted and corresponds with the patient’s visual acuity. The junction of ellipsoid zone disruption and loss is noted (green arrows). |
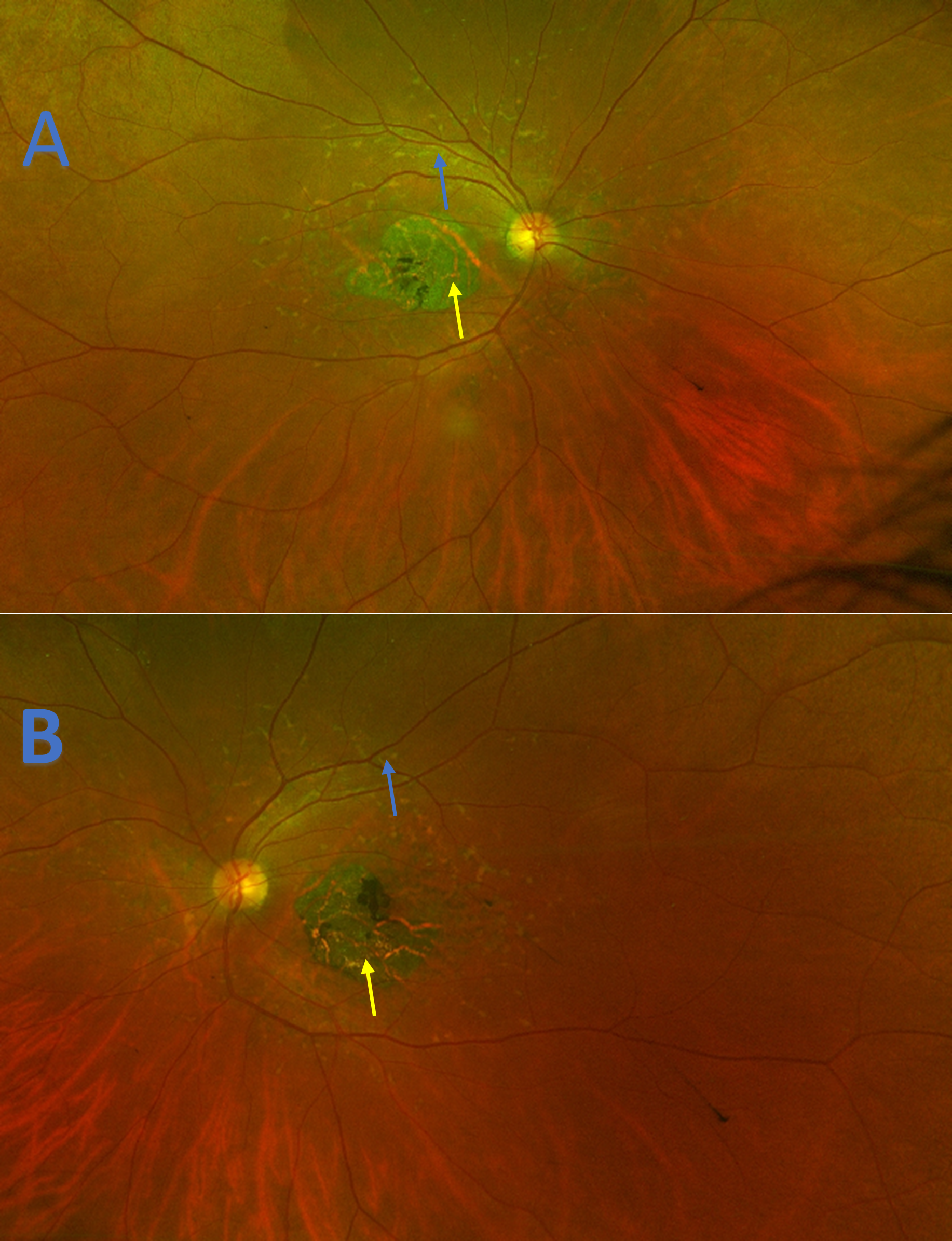 Figure 6. Ultra-widefield imaging demonstrating retinal pigment atrophy (yellow arrows) in the right eye (A) and left eye (B) of patient 2 that has progressed temporally when compared to initial presentation (Figure 4). Large amounts of pisciform fleck are observed extending into the arcades of both eyes (blue arrows). |
 Figure 7. Fundus autofluorescence of the right (A) and left eye (B) of patient 2 showing large areas of central macular hypo-autofluorescence (yellow arrows) corresponding to the macular atrophy observed in Figure 6. Mixed hyper- and hypo-autofluorescence (blue arrows) is noted surrounding the macular atrophy representing active lipofuscin accumulation (hyper) and reabsorbed lipofuscin/RPE atrophy (hypo). Peripapillary sparing is also visible in both eyes (blue circles). Generalized retinal pigment epithelium involvement as indicated by global hyper-autofluorescence is seen around the macular hypo-autofluorescence (yellow circles). |
 Figure 8. Montage progression images of the right eye (B, D, F) and left eye (A, C, E) for patient 2. Visit one (baseline) fundus images (A, B) show macular atrophy with compromise of the fovea (yellow arrows). Visit two fundus imaging (C, D) shows expansion of the macular atrophy with adjacent retinal pigment epithelium hyperplasia (yellow arrows) that now involves the fovea. This corresponds to the decrease in the patient’s visual acuity from visit one. Fundus autofluorescence (E, F) further highlights the macular atrophy from visit two as evident by the large amounts of hypo-autofluorescence (yellow arrows). Click to enlarge |
Education Guidelines
Key concepts
- The pathophysiology and specific genetics of STGD1
- Importance of multimodal imaging in STGD1
- Current and future treatment options for patients with STGD1
Learning objectives
- Define and recognize the varying clinical presentations of STGD1
- Identify the possible signs and symptoms associated with STGD1
- Discuss the possible differential diagnoses for STGD1
- Recognize the importance of utilizing additional testing to aid in diagnosing and managing STGD1
- Staying current and informed about novel research and potential treatment options
Discussion points
1. Knowledge, understanding and facts about the case and condition presentation
- Describe the typical appearance and presentation of early onset STGD1
- Describe the typical appearance and presentation of late onset STGD1
- Compare and contrast the pathogenesis and genetics of these two subtypes of STGD1
2. Differential diagnosis
- What other conditions present in a similar clinical manner to STGD1?
- How can a clinician differentiate between these similar conditions based on patient presentation (history, signs and symptoms) and multimodal imaging results?
3. Patient management and role of the optometrist
- What additional testing should be ordered for patients, and their family, who potentially have STGD1?
- Understand what referrals should be made for patients who present with visual acuity/visual field loss
4. Critical-thinking concepts
- Understand the varying phenotypes of STGD1 and how genotype influences this presentation
- Realize the potential of current research in regard to treatment options
Assessment of learning objectives
STGD1 is a complex disease owing to its complicated genotypic-phenotype interaction. Therefore, these teaching case reports are appropriate for third- and fourth-year students, as well as residents, who already have familiarity and knowledge of basic retinal anatomy, genetics and multimodal imaging interpretation. Suggested assessments include:
- Presenting the cases and discussion points as a component of a journal club at a student’s optometry school, optometric rotation site or residency site. The students and/or residents could work in groups or independently to answer the discussion questions.
- In a classroom setting, students could be presented the cases with the goal of formulating a final diagnosis and proper management plan.
- Comprehension and knowledge could be further evaluated through open-ended questioning to the class or through formal testing with multiple-choice questions. Comments and further expansion on tested concepts can be provided if the question is answered correctly or incorrectly. This would allow students to build upon their current knowledge base.
Discussion
On a genetic level, STGD1 is caused by mutations to the ABCA4 gene, which encodes for the transmembrane ABCA4 protein, an ATP binding cassette transporter that is exclusively found in the rim of the rod and cone outer segment discs.2 From the photoreceptors it transports retinoids to the RPE. More specifically, during the visual cycle, all-trans retinal is converted to 11-cis retinal. Light-activated rhodopsin and opsin release all-trans retinal, which combines with phosphatidylethanolamine to form N-retinylidene-phosphatidylethanolamine (N-ret-PE). N-ret-PE is typically transported to the photoreceptor’s disc surface for removal via the ABCA4 protein.1,2 In STGD1, the malfunctioning ABCA4 protein causes the retinoid compounds to accrue in the outer segments of photoreceptors, leading to toxic levels of phosphatidylpyridinium bisretinoid (A2PE) in the photoreceptor membranes. A2PE is then hydrolyzed to A2E, an extremely toxic metabolite and component of lipofuscin in RPE cells. Toxic/excessive levels eventually amass, which results in RPE damage and atrophy along with adjacent photoreceptor damage and loss.1,2,9
Other variations of Stargardt disease, due to autosomal dominant mutations, exist but to a much lesser extent than STGD1. One such mutation is found in the ELOVL4 (Stargardt disease 3, STGD3) gene on chromosome 6q16. ELOVL4 is fundamental for the proper synthesis of very long chain polyunsaturated fatty acids (VLC-PUFA). VLC-PUFA constitute a large part of the phosphatidylcholine that is found in the outer segments of both rods and cones.10,11 While the exact role of VLC-PUFA in the retina is unknown, it is believed to play an important role in photoreceptor membrane fluidity while also being potentially involved with phototransduction.11 This mutation(s) in ELOVL4 results in a deficiency of VLC-PUFA that eventually results in retinal degeneration that damages the RPE cells.11 Stargardt disease can also be inherited in an autosomal dominant fashion due to mutations in the PROM1 gene (Stargardt disease 4; STGD4). This gene has been implicated in the synthesis of photoreceptors with mutations leading to improper migration of PROM1 into photoreceptor outer segments with subsequent photoreceptor and RPE damage.10 Discussion of STGD3 and STGD4 are beyond the scope of this article; therefore, STGD1 is the focus.
As mentioned previously, mutations in ABCA4 are implicated in STGD1. ABCA4 is considered a large and complex gene thus mutations can present with diverse phenotypic appearances, even among STGD1 patients.1 Genetic and environmental factors are also thought to influence the phenotypic presentation as even family members with the same mutation can present differently.2 Mutations in the gene have been implicated in cone-rod dystrophies, retinitis pigmentosa and age-related macular degeneration (AMD).2,9,12 Genotype correlations to phenotypic presentation can be difficult owing to this heterogeneity. Overall, mutations can be classified into null mutations (which cause total loss of product encoded by gene) or missense mutations (alteration of the product encoded by the gene).5 Patients with null mutations tend to present with early onset cases of STGD1. They correspondingly have more severe presentations and a poorer overall prognosis. In contrast, patients with missense mutations have symptoms that correlate to a later disease onset with milder clinical signs and a better visual prognosis.13
Early clinical signs of STGD1 may include a normal appearing fundus that progresses to bilateral loss of the foveal reflex and/or granularity or mild disruption of the RPE. This would correlate to normal or mild vision loss.14 Fishtail or pisciform flecks can then develop within the posterior pole. These characteristic white-yellow lesions are composed of lipofuscin and are found at the level of the RPE.2,12,15 They can vary overtime being that they may remain, resolve and eventually result in RPE atrophy and significant visual acuity loss depending on their location. As the condition continues to progress, RPE and outer retinal damage occur leading to RPE and sensory retinal atrophy, sometimes termed a “beaten bronze” appearance.7 Further atrophy can in some cases result in a characteristic “bull’s eye” maculopathy, which would correlate to severe vision loss and likely central scotomas. Severe cases can show retinal atrophy extending into the arcades.7,16 Other findings can include a characteristic peripapillary sparing with STGD1. The reason for this is not fully understood. It is hypothesized that there may be an ideal ratio between the photoreceptors and RPE at this area and/or that the peripapillary area is less susceptible to photo-oxidative damage and lipofuscin accumulation owing to a thicker peripapillary retinal nerve fiber layer.17-19 Late onset STGD1 can present similarly but with milder findings. The fovea tends to be spared or mildly involved leading to better visual outcomes.20 It is unknown why the fovea is generally spared, but it is thought to be due to possible missense mutations.5
At times, the terms STGD1 and FF are used interchangeably; however, clinicians should note that they represent different phenotypic presentations with similar genotypes. FF can present in an equivalent manner to STGD1 but with more extensive retinal involvement with flecks extending into the midperiphery and a later disease onset.1,21 The fovea tends to be less involved, which may allow for better visual acuity/outcomes.21
This phenotypic heterogeneity was evident with our two patients. Patient 1 likely had early onset STGD1, with findings isolated to the macula, resulting in significant macular atrophy and subsequent visual acuity loss. In contrast, patient 2 likely had FF as indicated by the extension of the pisciform flecks into the midperiphery. The preservation of fairly good visual acuity until middle age also supports this diagnosis. Also, the patient having one missense mutation and no null mutations correlates to her later disease onset and initially milder clinical signs.13 It should be noted though, that patient 2 could have also used eccentric viewing to achieve her relatively good entering visual acuity. Eccentric viewing is a common technique used by patients with damage to the macula. By using this strategy patients can direct their eye so that the image falls on a non-foveal point or preferred retinal locus (PRL).22 PRLs can be determined using microperimetry. Unfortunately, this test was not available to us.
Grading systems
Due to these heterogeneities, various attempts have been made to aid clinicians in grading and classifying Stargardt disease. The grading systems differ in that they are based on fundus signs, genetic testing and results of multimodal imaging such as FAF. The Fishman system was developed by Dr. Gerald Fishman and categorizes Stargardt disease based on funduscopic appearance and electrophysiological testing.23 Stage 1 is characterized by macular pigmentary changes including mottling within 1 disc diameter of the fovea. Stage 2 shows pigmentary changes or flecks that extend into and beyond the arcades and/or nasally to the disc. Stage 3 shows RPE and choriocapillaris atrophy secondary to reabsorption of the flecks. Stage 4 is a continuation of stage 3 with worsening atrophy. Both patients in this case report would be classified as stage 4 based on this system. Genotype classification is based on the type and degree of underlying genetic mutations. Category A is for patients with two or more null mutations while category B includes patients with one severe null mutation and one or more missense mutations. Category C is patients with no null mutations but two or more missense mutations while category D is patients with one missense mutation.24 While genetic testing was not performed on patient 1, patient 2 would be in category D based on her single missense mutation. Other grading systems also exist based on FAF and electroretinogram (ERG) findings as well as a classification system for early onset Stargardt disease.16,24
Multimodal imaging
Although cases of STGD 1 may be obvious on initial presentation, many cases are not so apparent. As mentioned previously, early onset cases present without obvious fundus signs. One study noted that a quarter of children with STGD1 present with no obvious retinal lesions.25 Unfortunately, this can lead to delayed diagnosis. Research has revealed a median delay in diagnosis of 3 years.25 Misdiagnosis is not limited to early onset cases. Late-stage findings in STGD1 include RPE atrophy, which may be mistaken for atrophy seen in late stages of AMD. This underscores the need for multimodal imaging to aid in timely and correct diagnosis, which may allow for prompt referral for LVR and possible genetic treatment trials. A multitude of testing options are available to clinicians to help aid in the diagnosis and monitoring of STGD1. This includes traditional fundus photography, ultra-widefield fundus photography, fluorescein angiography, FAF and electrophysiology testing.
Fundus photographs allow for documentation of retinal findings such as RPE atrophy and/or pisciform flecks, which were easily visible in both patients. However, it is lacking in that it can only detect more obvious or superficial lesions. Nonetheless, it allows for long-term monitoring of patients and can be used as a patient education tool. Clinicians should perform fundus photography on all patients but, if available to them, should specifically consider ultra-widefield imaging/photography. This allows for the detection of not only central presentations but also peripheral presentations of STGD1 such as the extension of flecks observed in patient 2.5
Fluorescein angiography has been used traditionally to detect the so-called dark choroid finding that is present in a majority (up to 80%) of STGD1 cases.7 The dark choroid represents lipofuscin accumulation in the RPE, which results in the blockage of normal choroidal fluorescence.7,26 Although fluorescein angiography has its purposes for early and mild cases of STGD1, it is not as valuable for advanced stages as testing will reveal large amounts of RPE atrophy/window defects correlating to the absence of the dark choroid finding.2
By far the most useful test available to clinicians is FAF. As explained previously, STGD1 is characterized by the accumulation of lipofuscin, which leads to classic findings such as pisciform flecks and RPE atrophy. Lipofuscin has autofluorescent properties and thus FAF can visualize/detect these findings despite the appearance of a normal fundus. Two characteristic findings are seen on FAF: hyper-autofluorescence and hypo-autofluorescence. Hypo-autofluorescence indicates a lack of RPE and thus a lack of lipofuscin, which correlates to RPE atrophy that can be seen in STGD1. Hyper-autofluorescence indicates an excess of lipofuscin in the RPE and thus a diseased or sick RPE. This also correlates to pisciform flecks of various sizes and shapes. Ultimately, the hyper-autofluorescence seen with pisciform flecks and within the RPE will transition to hypo-autofluorescence as the flecks and lipofuscin are reabsorbed leading to RPE atrophy. Other findings are also present with FAF. One finding that can be seen in patients is a hyper-autofluorescent border surrounding RPE atrophy/hypo-autofluorescence. This is thought to indicate possible disease progression in terms of RPE atrophy. FAF patterns have also been classified into subtypes based on the degree of atrophy. Subtype 1 is thought to represent the mildest form with slower rates of progression while the last subtype, 3, is associated with more rapid progression.16 Lastly, peripapillary sparing is more evident and obvious with FAF when compared to fundus examination/photography.2 These FAF findings were apparent in both patients as they presented with central hypo-autofluorescence, surrounding hyper-autofluorescent flecks, a hyper-autoflourescent border around the hypo-autofluorescence, and peripapillary sparing (Figure 7). Clinicians should perform baseline FAF in all patients that have or are suspected of having STGD1 based on the findings above. Baseline FAF should also be considered in family members if history or examination findings suggest possible familial involvement.
SD-OCT serves as a vital tool for assessing specific areas of retinal and choroidal involvement and for monitoring disease progression. The earliest known finding in children that has been imaged with OCT is external limiting membrane thickening.27,28 However, these patients may be asymptomatic at such a young age making its detection more difficult. As STGD1 is a disease of the outer retina, the inner retinal layers will be spared thus typical findings include damage and disruption to the macular outer retina layers including the EZ and the RPE. As the condition progresses, total atrophy of the layers will occur with preservation of the more peripheral macular tissue. These findings were seen in both patients as patient 1 had central EZ loss with accompanying RPE atrophy. Her peripheral retina was spared of any damage. Patient 2 had more mild initial findings as seen with foveal EZ and RPE disruption. However, this eventually transitioned to more EZ and RPE involvement and vision loss. This correlates to the typical bull’s eye maculopathy appearance that is associated with STGD1. As almost all commercially available SD-OCT instruments have progression software, clinicians can use specific parameters such as total retinal thickness, macular volume and outer retinal thickness to monitor progression.1 Although not needed to diagnose or monitor the condition, newer types of OCT, such as enhanced depth OCT and swept source OCT, can be used to visualize and image the choroid and its possible role in STGD1. Researchers have described four choroidal patterns that correlate with the loss of retinal structures and retinal integrity: stage 1 represents a normal choroid; stage 2 represents a reduced Sattler or Haller layer; stage 3 represents a reduced Sattler and Haller layer; stage 4 represents reduced Sattler and Haller layers with choroidal caverns.4 Currently, investigators are unsure why these caverns form. Enhanced depth imaging was not performed in either patient involved in this case report. However, based on the appearance of the choroid on the available SD-OCT scans, patient 1 likely had stage 2 choroidal involvement with a reduced Sattler layer while patient 2 likely had stage 3 choroidal involvement.
Newer imaging technologies such as optical coherence tomography angiography (OCT-A) are emerging as possible imaging options for clinicians. Although FAF and SD-OCT are considered better imaging modalities, OCT-A has the benefit of allowing imaging of the retinal and choroidal microvasculature. Although they are a rare finding in STGD1, OCT-A could aid in diagnosing choroidal neovascular membranes that may not be visible with SD-OCT and/or FAF.29 OCT-A has also allowed for a better understanding of the pathophysiology of STGD1. Research suggests that RPE damage on FAF is larger than choriocapillaris damage on OCT-A thus providing evidence for initial RPE involvement followed by choroidal damage.4
Functional testing including visual field testing can also be used in the monitoring of STGD1 patients. As the condition is mild in the early stages due to limited RPE and outer retinal damage, formal testing with static and kinetic perimetry is usually normal. As the condition progresses secondary to greater amounts of RPE involvement, relative central scotomas can be detected with either testing modality. Eventually, with greater amounts of RPE damage, patients transition to absolute central scotomas.21,30 Exceptions can include patients with spared foveas, which correlates to ring scotoma-like field defects.31 Although not traditionally associated with STGD, peripheral visual field loss can occur in patients whose disease has spread to the retinal periphery as evidenced by ultra-widefield imaging.32 As noted previously, microperimetry can also be used in the management of STGD1 as its built-in fundus tracking and fixation monitoring enable PRL measurements. This allows clinicians to quantify a patient’s remaining visual function.2 Unfortunately, this technology is not readily available to all clinicians; the devices tend to be located at larger clinical research sites or LVR clinics. This underscores the need to refer patients to such sites when appropriate. Although visual field testing was not performed in either patient in this case report, the amount and degree of retinal atrophy seen in both patients would likely have resulted in absolute scotomas.
Next, electrophysiological testing can aid in the diagnosis of STGD1 but may be better suited for helping to predict patients’ prognosis. Pattern ERG and focal ERG signals are typically not present or are noticeably reduced in patients with STGD1. This would correlate to macular involvement and damage; however, this might be also noted on OCT and FAF.33 What may be of more importance to clinicians is the classification system based on electrophysiological findings created by Lois et al. They categorized subjects into 3 groups: group 1 – severe pattern ERG but normal scotopic and photopic ERG; group 2 – loss of photopic function; group 3 – additional photopic disfunction along with scotopic disfunction.34 With this classification system in place, another study demonstrated that patients with ERG patterns in group 1 had the best visual prognosis while patients in groups 2 and 3 had intermediate and poor prognoses, respectively.24
Differential diagnosis
Different combinations of ABCA4 mutations are predicted to result in distinct phenotypes. Evidence indicates that ABCA4 is involved in the development of various other retinal diseases beyond STGD1, such as AMD, cone-rod dystrophies and rod-cone dystrophies such as retinitis pigmentosa. Therefore, these phenotypic variations should be considered as differentials before a final diagnosis is confirmed. Other common differentials are:
- AMD. AMD tends to present later in life and is characterized by the presence of drusen, which may be confused for lipofuscin flecks, in its dry form and the presence of choroidal neovascular membranes in its wet form. The flecks and drusen can be distinguished with FAF as the flecks will show intense hyper-autofluorescence while the drusen will show little to no autofluorescence.12 Both conditions tend to present with RPE atrophy in their respective late stages; however, this presentation should be evident at an earlier age in STGD1 patients. AMD patients may also have a strong history of smoking and/or exposure to ultraviolet light.
- Cone-rod dystrophies/rod-cone dystrophies. Patients with a cone-rod dystrophy have an obvious lack of color vision but may also have foveal atrophy as seen in STGD1 patients. They lack the flecks (either on FAF or fundus examination) seen in STGD1 patients. The fundus presentation in rod-cone dystrophies shows bone spicules, vessel attenuation and optic disc pallor, features that are not seen in typical STGD1. Nyctalopia is also associated with these patients, a symptom that is rarely seen with STGD1 patients.
- Pattern dystrophies. These are a set of conditions that manifest at the macula in middle age and include adult onset vitelliform dystrophy, butterfly-shaped pigment dystrophy, reticular dystrophy, multifocal pattern dystrophy simulating Stargardt disease, and fundus pulverulentus. Most of the patients present with only mild vision loss with little or slow progression. The pattern dystrophies are due to mutations in the PRPH2 gene, which can be confirmed through genetic testing. The conditions also lack a dark choroid that can be seen in most STGD1 patients.12
- Pentosan polysulfate sodium (Elmiron) toxicity. Pentosan polysulfate is a medication commonly used in the treatment of interstitial cystitis but recently has been linked with a maculopathy that presents similarly to STGD1 based on fundus presentation and FAF patterns. A history of chronic pentosan polysulfate use can support the diagnosis.35
Treatment
As previously mentioned, no treatment for STGD1 is currently available.10 However, gene replacement, stem cell therapy and pharmacological approaches, specifically vitamin A visual cycle modulators, are being investigated in clinical trials.
The objective of gene therapy is to introduce a properly functioning gene into the retina to account for the missing or mutated gene in question. This can be done either through intravitreal injections or through subretinal delivery of the gene.2 This treatment option has been effective as evidenced by the many current clinical trials for inherited disease with one approved gene therapy on the market, voretigene neparvovec-rzyl (Luxturna).36 Gene therapy could potentially be applied to STGD1; however, the size of the ABCA4 gene (6.4 kb) presents a problem. Most human gene therapy has used adeno-associated virus (AAV) vectors, which have a capacity of approximately 4.7 kb.5,9 In hopes of overcoming this problem, researchers have shifted toward dual AAV vector delivery in which a gene is split and placed in two separate AAV vectors. Studies at this time have been limited to preclinical trials.9 An alternative is lentiviral vectors which have a larger capacity (8 kb) to house the ABCA4 gene. Unfortunately, studies have shown little efficacy with this method.2
Stem cell therapy is an additional future treatment option. In a phase 1/2 clinical trial, STGD1 patients received RPE cells derived from human embryonic stem cells.14,18 The study showed no safety concerns but was too small to determine efficacy.37 A similar study was performed in which patients received escalating doses of human embryonic stem cells/RPE cells. Results were similar to the first study with no adverse effects but also no improvement in retinal function or vision.38 Follow-up studies are ongoing.9 Clinicians should also consider whether stem cell transplantation is a viable long-term option as it does not address the actual cause of the disease, the gene mutation. These stem cells are possibly a temporary option; however, they themselves can possibly still accumulate A2E and other toxic molecules.39
Other treatment possibilities under investigation are pharmacological agents or visual cycle modulators. These target different components of the visual cycle in hopes of preventing the accumulation of toxic molecules in the RPE.2 One such agent is emixustat, which inhibits retinol isomerase RPE 65 leading to the reduced availability of 11-cis- and all-trans-retinal, and eventually toxic A2E.2 Another option being studied is a deuterated vitamin A (ALK-001). In this form of vitamin A, hydrogen atoms are replaced with deuterium atoms. This substitution impedes vitamin A dimerization and thus reduces production of A2E.2,9 Fenretinide is also being examined as a treatment option. It is a synthetic derivative of vitamin A that binds to and reduces free circulating retinol binding protein. This complex is excreted in the urine leading to decreased levels of vitamin A and thus A2E.2,9
While vitamin A alternatives are being studied, clinicians should also keep in mind the role of natural vitamin A supplementation in STGD1 patients. Excess intake of vitamin A is thought to lead to accelerated disease progression in some STGD1 patients as it leads to increased dimerization and thus lipofuscin formation.40 However, an evidence-based review of vitamin A supplementation in STGD1 patients concluded that there are very few large studies from which to draw solid conclusions.15 At this time, the National Eye Institute and the National Institutes of Health still advise patients to avoid supplements that contain more than the daily recommend allowance of vitamin A.41
RPE atrophy in STGD1 is due to A2E and other bisretinoids; however, evidence suggests that these compounds also activate the complement system leading to further atrophy.2 Complement activation has also been implicated heavily in the development and progression of RPE atrophy in AMD patients.2,42 Specifically, overactivation of the C5 and C3 components of the complement system leads to eventual activation of a membrane attack complex (MAC). MAC activation then leads to cell lysis and RPE and photoreceptor atrophy. Therefore, inhibition of C5 and C3 could lead to downstream inhibition of MAC and decreased RPE atrophy.43 Due to the similar features of the conditions, therapies designed for AMD may be applicable to STGD1. Two such recently FDA-approved medications are pegcetacoplan (Syfovre), a C3 inhibitor, and avacincaptad pegol (Izervay), a C5 inhibitor. Results from trials of both drugs have been promising in terms of preventing RPE progression in AMD.44-46 However, indications for STGD1 are still under investigation. A phase 2b clinical study, the STAR trial, is ongoing for Izervay in the treatment of STGD1.47
Dietary supplementation for the treatment of STGD1 disease is also being studied. Omega-3 fatty acids including docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA) and alpha-linolenic acid (ALA) are being investigated to determine whether they can improve macular function. A recent study showed improvement in objective and subjective vision in AMD and STGD1 patients using omega-3s.48 Although promising, further research is needed as this study was limited to 21 patients.
With so many treatment options being investigated, the newest option may provide the best outcomes. Clustered regularly interspaced short palindrome repeat (CRISPR) is gene editing technology that can correct for gene errors.49 The technology has been demonstrated to repair DNA in mice with genetic disorders and has the potential to treat varied inherited retinal disorders.50 Currently, studies are lacking in human subjects.
Until a proven treatment option exists for STGD1 patients, clinicians should consider rehabilitative options, mainly LVR, to improve or maintain a patient’s quality of life.51 LVR can allow patients to resume and continue to perform activities of daily living such as reading, writing and driving with the assistance of hand magnifiers, telescopes and electronic devices.52 Patients can be followed with multimodal imaging yearly to monitor for disease progression while waiting on potential treatment options. However, until that time, LVR services are vital to maintain quality of life. Although both patients in this case report were educated on the benefits of LVR, they ultimately did not receive these services as one patient was lost to follow-up and the other declined. The decision to seek LVR is ultimately the patient’s; however, clinicians should continue to advocate for it even when patients think the services are not necessary.
It is also recommended that patients with STGD1 and patients who are suspected to have STGD1 undergo genetic testing and receive genetic counseling.10,16 Genetic testing can help confirm and refine the diagnosis and allow patients to better understand how STGD1 can affect their vision throughout life.16 Genetic testing can also help individuals qualify for certain clinical trials and future treatment options.23
Conclusion
STGD1 is a highly complex disease based on its phenotypic and genotypic heterogeneity.2 It can manifest with variable signs and symptoms. Therefore, multimodal imaging and genetic testing play an important role in arriving at a proper diagnosis. Although no treatment exists currently, research into potential therapies is promising. Until an effective treatment emerges, clinicians should continue to monitor their patients for disease progression and make appropriate referrals for LVR.
References
- Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol. 2017 Jan;101(1):25-30. doi: 10.1136/bjophthalmol-2016-308823.
- Cicinelli MV, Battista M, Starace V, Battaglia Parodi M, Bandello F. Monitoring and management of the patient with Stargardt disease. Clin Optom (Auckl). 2019 Nov 28;11:151-165. doi: 10.2147/OPTO.S226595.
- Stargardt’s disease [Internet]. Victoria, Australia: Centre for Eye Research Australia; c2023 [updated 2020-08-20]. Available from: https://www.cera.org.au/conditions/stargardts-disease/.
- Heath Jeffery RC, Chen FK. Stargardt disease: multimodal imaging: a review. Clin Exp Ophthalmol. 2021 Jul;49(5):498-515. doi: 10.1111/ceo.13947.
- Huang D, Heath Jeffery RC, Aung-Htut MT, et al. Stargardt disease and progress in therapeutic strategies. Ophthalmic Genet. Feb 2022;43(1):1-26. doi:10.1080/13816810.2021.1966053.
- Sun H, Smallwood PM, Nathans J. Biochemical defects in ABCR protein variants associated with human retinopathies. Nat Genet. Oct 2000;26(2):242-6. doi:10.1038/79994.
- Tsang SH, Sharma T. Stargardt disease. Adv Exp Med Biol. 2018;1085:139-151. doi:10.1007/978-3-319-95046-4_27.
- Westerfeld C, Mukai S. Stargardt’s disease and the ABCR gene. Semin Ophthalmol. Jan-Feb 2008;23(1):59-65. doi:10.1080/08820530701745249.
- Piotter E, McClements ME, MacLaren RE. Therapy approaches for stargardt disease. Biomolecules. 2021 Aug 9;11(8):1179. doi: 10.3390/biom11081179.
- Imani S, Cheng J, Shasaltaneh MD, et al. Genetic identification and molecular modeling characterization reveal a novel PROM1 mutation in Stargardt4-like macular dystrophy. Oncotarget. 2017 Nov 9;9(1):122-141. doi: 10.18632/oncotarget.22343.
- Donato L, Scimone C, Rinaldi C, et al. Stargardt phenotype associated with two ELOVL4 promoter variants and ELOVL4 downregulation: new possible perspective to etiopathogenesis? Invest Ophthalmol Vis Sci. 2018 Feb 1;59(2):843-857. doi: 10.1167/iovs.17-22962.
- Kohli P, Kaur K. Stargardt disease. [Updated 2023 May 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK587351/.
- Simonelli F, Testa F, Zernant J, et al. Genotype-phenotype correlation in Italian families with Stargardt disease. Ophthalmic Res. 2005 May-Jun;37(3):159-67. doi: 10.1159/000086073.
- Fishman GA, Farber M, Patel BS, Derlacki DJ. Visual acuity loss in patients with Stargardt’s macular dystrophy. Ophthalmology. Jul 1987;94(7):809-14. doi:10.1016/s0161-6420(87)33533-x
- Federspiel CA, Bertelsen M, Kessel L. Vitamin A in Stargardt disease-an evidence-based update. Ophthalmic Genet. 2018 Oct;39(5):555-559. doi: 10.1080/13816810.2018.1488174.
- Fujinami K, Sergouniotis PI, Davidson AE, et al. Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am J Ophthalmol. 2013 Sep;156(3):487-501.e1. doi: 10.1016/j.ajo.2013.05.003.
- Nassisi M, Mohand-Saïd S, Andrieu C, et al. Peripapillary sparing with near infrared autofluorescence correlates with electroretinographic findings in patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2019 Dec 2;60(15):4951-4957. doi: 10.1167/iovs.19-27100.
- Cideciyan AV, Swider M, Aleman TS, et al. ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest Ophthalmol Vis Sci. Dec 2005;46(12):4739-46. doi:10.1167/iovs.05-0805.
- Brandstetter C, Mohr LK, Latz E, Holz FG, Krohne TU. Light induces NLRP3 inflammasome activation in retinal pigment epithelial cells via lipofuscin-mediated photooxidative damage. J Mol Med (Berl). Aug 2015;93(8):905-16. doi:10.1007/s00109-015-1275-1
- van Huet RA, Bax NM, Westeneng-Van Haaften SC, et al. Foveal sparing in Stargardt disease. Invest Ophthalmol Vis Sci. 2014 Oct 16;55(11):7467-78. doi: 10.1167/iovs.13-13825.
- Stargardt disease/fundus flavimaculatus [Internet]. EyeWiki, American Academy of Ophthalmology. Available from: https://eyewiki.aao.org/Stargardt_Disease/Fundus_Flavimaculatus.
- Costela FM, Kajtezovic S, Woods RL. The preferred retinal locus used to watch videos. Invest Ophthalmol Vis Sci. 2017 Dec 1;58(14):6073-6081. doi: 10.1167/iovs.17-21839.
- Fishman GA. Fundus flavimaculatus. A clinical classification. Arch Ophthalmol. Dec 1976;94(12):2061-7. doi:10.1001/archopht.1976.03910040721003.
- Fujinami K, Lois N, Mukherjee R, et al. A longitudinal study of Stargardt disease: quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Invest Ophthalmol Vis Sci. Dec 17 2013;54(13):8181-90. doi:10.1167/iovs.13-12104.
- Bax NM, Lambertus S, Cremers FPM, Klevering BJ, Hoyng CB. The absence of fundus abnormalities in Stargardt disease. Graefes Arch Clin Exp Ophthalmol. Jun 2019;257(6):1147-1157. doi:10.1007/s00417-019-04280-8.
- Lambertus S, van Huet RA, Bax NM, et al. Early-onset stargardt disease: phenotypic and genotypic characteristics. Ophthalmology. Feb 2015;122(2):335-44. doi:10.1016/j.ophtha.2014.08.032.
- Fujinami K, Singh R, Carroll J, et al. Fine central macular dots associated with childhood-onset Stargardt Disease. Acta Ophthalmol. Mar 2014;92(2):e157-9. doi:10.1111/aos.12259.
- Lee W, Nõupuu K, Oll M, et al. The external limiting membrane in early-onset Stargardt disease. Invest Ophthalmol Vis Sci. Aug 19 2014;55(10):6139-49. doi:10.1167/iovs.14-15126.
- Pawlak D, Souied E, Mimoun G, et al. Clinical features of choroidal neovascularization as complication of late-onset Stargardt disease. Invest Ophthalmol Vis Sci. May 2004;45(13):3079.
- Bernstein A, Sunness JS, Applegate CA, Tegins EO. Mapping the dense scotoma and its enlargement in Stargardt disease. Retina. 2016 Sep;36(9):1741-50. doi: 10.1097/IAE.0000000000001003.
- Kumar S, El Menaisi AM. Stargardt’s disease presenting with bilateral central ring scotoma. Saudi J Ophthalmol. Oct-Dec 2018;32(4):349-352. doi:10.1016/j.sjopt.2018.07.008.
- Abalem MF, Otte B, Andrews C, et al. Peripheral visual fields in ABCA4 Stargardt disease and correlation with disease extent on ultra-widefield fundus autofluorescence. Am J Ophthalmol. Dec 2017;184:181-188. doi:10.1016/j.ajo.2017.10.006.
- Stavrou P, Good PA, Misson GP, Kritzinger EE. Electrophysiological findings in Stargardt’s-fundus flavimaculatus disease. Eye (Lond). 1998;12 (Pt 6):953-8. doi:10.1038/eye.1998.247.
- Lois N, Holder GE, Bunce C, Fitzke FW, Bird AC. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch Ophthalmol. 2001 Mar;119(3):359-69. doi: 10.1001/archopht.119.3.359.
- Vora RA, Patel AP, Yang SS, Melles R. A case of pentosan polysulfate maculopathy originally diagnosed as stargardt disease. Am J Ophthalmol Case Rep. 2020 Jan 25;17:100604. doi: 10.1016/j.ajoc.2020.100604.
- Maguire AM, Bennett J, Aleman EM, Leroy BP, Aleman TS. Clinical perspective: treating RPE65-associated retinal dystrophy. Mol Ther. 2021 Feb 3;29(2):442-463. doi: 10.1016/j.ymthe.2020.11.029.
- Schwartz SD, Regillo CD, Lam BL, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: follow-up of two open-label phase 1/2 studies. Lancet. Feb 7 2015;385(9967):509-16. doi:10.1016/s0140-6736(14)61376-3.
- Mehat MS, Sundaram V, Ripamonti C, et al. Transplantation of human embryonic stem cell-derived retinal pigment epithelial cells in macular degeneration. Ophthalmology. 2018 Nov;125(11):1765-1775. doi: 10.1016/j.ophtha.2018.04.037.
- Parmar VM, Parmar T, Arai E, Perusek L, Maeda A. A2E-associated cell death and inflammation in retinal pigmented epithelial cells from human induced pluripotent stem cells. Stem Cell Res. Mar 2018;27:95-104. doi:10.1016/j.scr.2018.01.014.
- Sofi F, Sodi A, Franco F, et al. Dietary profile of patients with Stargardt’s disease and retinitis pigmentosa: is there a role for a nutritional approach? BMC Ophthalmol. 2016 Jan 22;16:13. doi: 10.1186/s12886-016-0187-3.
- Stargardt disease [Internet]. Bethesda, MD: National Eye Institute; [updated Sept 29, 2021]. Available from: https://www.nei.nih.gov/learn-about-eye-health/eye-conditions-and-diseases/stargardt-disease.
- Dhooge PPA, Runhart EH, Li CHZ, et al. Systemic complement activation levels in Stargardt disease. PLoS One. 2021;16(6):e0253716. doi:10.1371/journal.pone.0253716.
- Desai D, Dugel PU. Complement cascade inhibition in geographic atrophy: a review. Eye (Lond). 2022 Feb;36(2):294-302. doi: 10.1038/s41433-021-01765-x.
- Jaffe GJ, Westby K, Csaky KG, et al. C5 inhibitor avacincaptad pegol for geographic atrophy due to age-related macular degeneration: a randomized pivotal phase 2/3 trial. Ophthalmology. 2021 Apr;128(4):576-586. doi: 10.1016/j.ophtha.2020.08.027.
- Liao DS, Grossi FV, El Mehdi D, et al. Complement C3 inhibitor pegcetacoplan for geographic atrophy secondary to age-related macular degeneration: a randomized phase 2 trial. Ophthalmology. 2020 Feb;127(2):186-195. doi: 10.1016/j.ophtha.2019.07.011.
- Abidi M, Karrer E, Csaky K, Handa JT. A clinical and preclinical assessment of clinical trials for dry age-related macular degeneration. Ophthalmol Sci. 2022 Aug 19;2(4):100213. doi: 10.1016/j.xops.2022.100213.
- Zimura compared to sham in patients with autosomal recessive stargardt disease (STGD1) [Internet]. U.S. National Library of Medicine, ClinicalTrials.gov. Available from: https://classic.clinicaltrials.gov/ct2/show/NCT03364153.
- Prokopiou K, Kolovos P, Tsangari H, et al. A prospective, multicentre, randomised, double-blind study designed to assess the potential effects of omega-3 fatty acids supplementation in dry age-related macular degeneration or Stargardt disease. Invest Ophthalmol Vis Sci. 2022;63(7):377-F0208.
- Redman M, King A, Watson C, King D. What is CRISPR/Cas9? Arch Dis Child Educ Pract Ed. 2016 Aug;101(4):213-5. doi: 10.1136/archdischild-2016-310459.
- Kantor A, McClements ME, Peddle CF, et al. CRISPR genome engineering for retinal diseases. Prog Mol Biol Transl Sci. 2021;182:29-79. doi:10.1016/bs.pmbts.2021.01.024.
- Scott IU, Smiddy WE, Schiffman J, Feuer WJ, Pappas CJ. Quality of life of low-vision patients and the impact of low-vision services. Am J Ophthalmol. Jul 1999;128(1):54-62. doi:10.1016/s0002-9394(99)00108-7.
- Shah P, Schwartz SG, Gartner S, Scott IU, Flynn HW Jr. Low vision services: a practical guide for the clinician. Ther Adv Ophthalmol. 2018 Jun 11;10:2515841418776264. doi: 10.1177/2515841418776264.

