
PEER REVIEWED
Neuromyelitis Optica Spectrum Disorder and MOGAD Optic Neuritis
Raman Bhakhri, OD, FAAO, Christopher J. Borgman, OD, FAAO, and Leonard V. Messner, OD, FAAO
Abstract
Optic neuritis is an inflammatory disorder of the optic nerve secondary to infectious and noninfectious causes. In traditional thinking, 90% of optic neuritis is associated with demyelinating disease, namely multiple sclerosis (MS). However, with recent advances in testing and technology, two atypical conditions that result in demyelination and subsequent optic neuritis have been isolated and identified, neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD). Although the clinical signs and symptoms can overlap with MS, the overall clinical course, treatment options and outcomes with NMOSD- and MOGAD-associated optic neuritis can greatly vary. This is owing to a separate and distinct pathophysiology. We report two cases of atypical optic neuritis associated with these conditions with diagnosis based on clinical findings and additional testing. A comprehensive review of the conditions is also presented including pathophysiology and treatment options.
Key Words: neuromyelitis optica, myelin oligodendrocyte glycoprotein, optic neuritis, neuroimaging, multiple sclerosis
Introduction
Typical optic neuritis (ON) has historically been described as inflammatory demyelination, most commonly associated with multiple sclerosis (MS).1 However, a new era of biomarkers has expanded the identification of atypical causes of ON to include neuromyelitis optica spectrum disorder (NMOSD) and myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD).1 Historically, there has been significant discussion about whether MS, NMOSD and MOGAD are different entities or similar presentations of a same-spectrum disease.1,2 However, with the identification of these new biomarkers, it is now accepted that they are indeed separate entities.1-6 As prognosis and chronic treatment options vary among these conditions, eyecare practitioners need to be aware of typical clinical findings and available testing for biomarkers to help identify and differentiate these separate diseases.1,3,4 This case report highlights two cases of atypical ON, NMOSD ON and MOGAD ON, which were ultimately diagnosed with the aid of ancillary testing and multimodal imaging.
Case #1 Description
A 30-year-old Hispanic male presented with sudden, painful vision loss over 3 days in both eyes. He reported mild photophobia, pain on eye movement, and that vision in his right eye was worse than in his left eye. The patient reported he had presented to a local emergency department 2 days prior where computed tomography of his brain was found to be normal. However, he sought a second opinion at our clinic because his vision continued to decline. His ocular and medical history were unremarkable. Medications included acetaminophen 500 mg as needed. He had no known allergies to medications and his social history was unremarkable. Entering visual acuity was 20/800 in the right eye and 20/125 in the left eye, with no improvement with pinhole testing. Pupils were equal in size, round and responsive to light. Swinging flashlight test revealed a right relative afferent pupillary defect (RAPD). Extraocular muscle motility was full in both eyes, but the patient reported pain with eye movement in all gazes. Slit lamp examination was unremarkable, and intraocular pressure (IOP) measured 12 mmHg in each eye with Goldmann applanation tonometry. Dilated fundus exam revealed bilateral ON edema (Figure 1). The macula and peripheral retina were unremarkable in both eyes. Threshold 30-2 visual field testing was performed and showed overall depression in both eyes (Figure 2). Baseline spectral-domain optical coherence tomography (OCT) of the optic nerves was obtained. A large amount of retinal nerve fiber layer (RNFL) thickening was noted in the right eye, more so along the superior and inferior portions of the optic nerve head. Thickening was also noted inferiorly in the left eye. Unfortunately, the data for the superior portion of the left optic nerve was not reliable due to scanning artifact (Figure 3). Emergency magnetic resonance imaging (MRI) revealed bilateral optic nerve enhancement consistent with bilateral ON (Figure 4). Given the bilateral ON findings and entering visual acuities, NMOSD was suspected and confirmed with positive aquaporin-4 immunoglobulin G (AQP4-IgG) titers.
 Figure 1. Fundus images of the right (A) and left eye (B) showing mild to moderate disc edema. The edema is noted to be greater in the right eye. Click to enlarge |
 Figure 2. Humphrey visual field testing (30-2 protocol) showing severely depressed visual fields in both eyes, with greater depression in the right eye. This corresponds with the more severe initial presentation of the right eye (Figure 1) and with the larger amounts of retinal nerve fiber layer thickening seen in Figure 3. Click to enlarge |
 Figure 3. Retinal nerve fiber layer (RNFL) scans of the right and left optic nerves. Significant thickening is present in the right eye, especially in the superior and inferior portions of the nerve. Thickening is also noted in the inferior portion of the left nerve with artifact (red arrows) preventing proper imaging of the superior portion of the nerve. The artifact corresponds to artificial RNFL thinning superiorly (purple arrow). Click to enlarge |
 Figure 4. A: Orbital axial magnetic resonance imaging (MRI) without contrast showing posterior optic nerve hyperintensities (yellow arrows) suggestive of bilateral optic neuritis. B: Orbital axial MRI with contrast and fat suppression showing extensive bilateral enhancement of the optic nerves (yellow arrows) confirming the diagnosis of bilateral optic neuritis. Click to enlarge |
The patient underwent intravenous methylprednisolone treatment and was prescribed chronic treatment with mycophenolate mofetil (MMF), which resulted in a full recovery of his vision. Unfortunately, the patient has been unable to follow-up in the clinic due to travel issues. He is currently monitored by an outside neurologist. In a phone conversation, the patient reported that with the prescribed treatment he had no NMOSD recurrence in approximately 3 years.
Case #2 Description
A 51-year-old African American woman presented complaining of painful, sudden vision loss in her right eye approximately 1 week earlier. The pain was noted upon eye movement in all gazes. She stated that her vision was improving since the initial onset of vision loss. Medical history was unremarkable with no known allergies. Entering visual acuity was 20/20- in the right eye and 20/20 in the left. Trace RAPD was observed in the right eye with corresponding 10% dyschromatopsia as measured by red cap desaturation. Extraocular muscle motility was full in both eyes. Confrontation visual field test results were normal in each eye. External ocular exam and slit lamp exam were unremarkable, and IOP measured 14 mmHg in each eye with Goldmann applanation tonometry. Dilated fundus examination revealed an essentially normal appearance of both optic nerves with a cup to disc ratio of 0.45/0.45 in the right eye and 0.35/0.35 in the left eye. OCT RNFL scans revealed one clock hour of borderline thinning inferior-temporal in the right eye and one clock hour of severe thinning inferior-temporal in the left eye. Ganglion cell complex (GCC) analysis showed generalized 360-degree thinning in the right eye with mild inferior-temporal thinning in the left eye (Figure 5). Retrobulbar ON was suspected, and MRI of the brain with and without contrast was performed and found to be essentially normal. Orbital imaging was not obtained. The patient was diagnosed with idiopathic retrobulbar ON and asked to return for follow-up in 3 to 4 months.
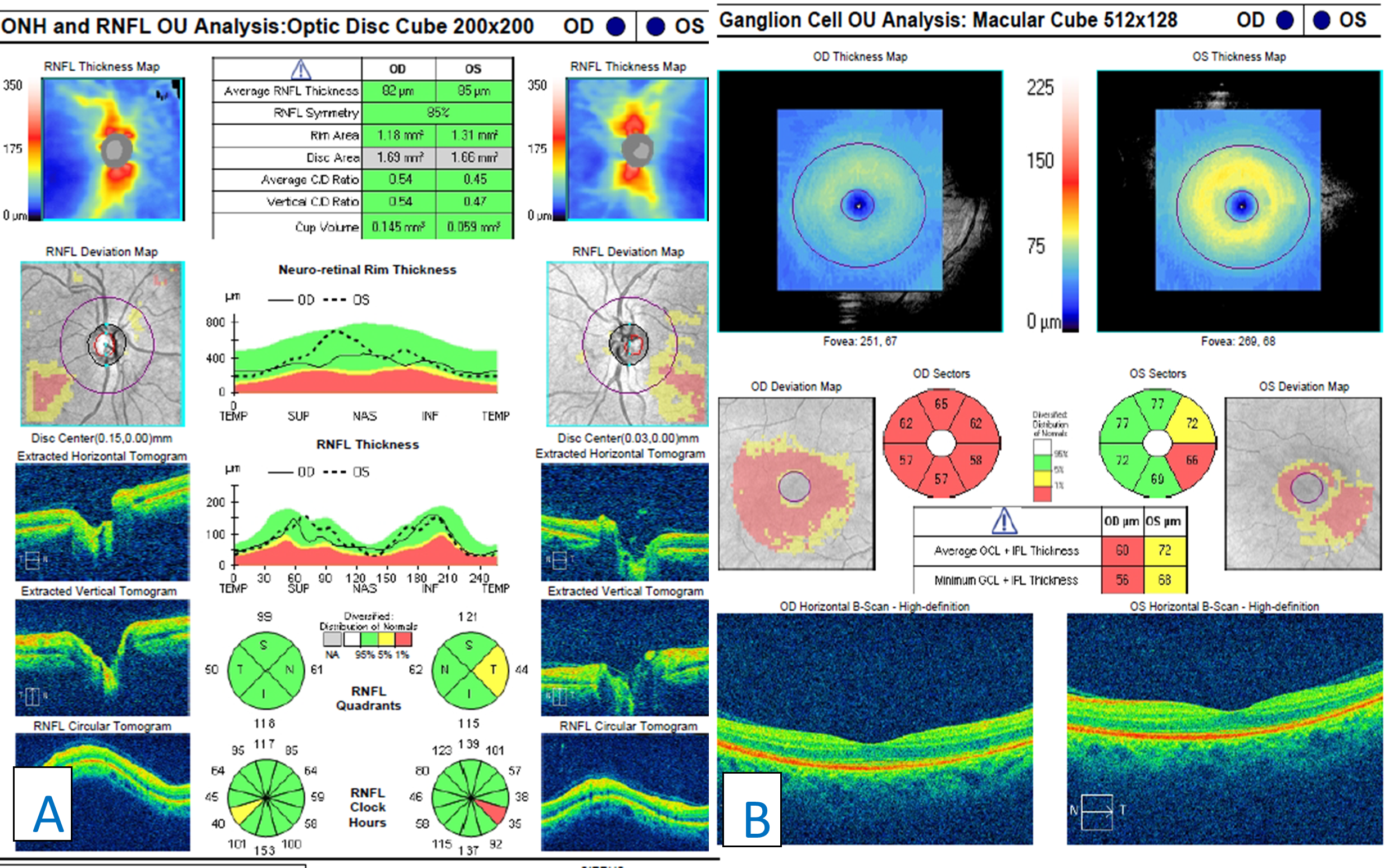
Figure 5. Retinal nerve fiber layer scans of both optic nerves showing one clock hour of borderline inferior-temporal thinning in the right eye and one clock hour of severe inferior-temporal thinning in the left eye (A). Ganglion cell complex analysis showing generalized 360-degree thinning in the right eye with mild inferior-temporal thinning in the left eye (B). Click to enlarge
The patient was lost to follow-up but presented again 7 years later complaining of similar painful vision loss, this time in the left eye, for 1 to 2 weeks. Her entering visual acuity was 20/20 OD and 20/60 OS with a 1+ RAPD in the left eye. Entrance testing was otherwise unremarkable. Slit lamp examination was also unremarkable. IOP measured 19 mmHg in the right eye and 18 mmHg in the left eye. Dilated fundus examination revealed generalized optic disc pallor in the right eye. The pallor corresponded to the patient’s history of ON. Examination of the left eye revealed an edematous and hyperemic optic nerve. Cup to disc ratios were stable compared with previous exams. OCT raster scans showed a flat optic nerve in the right eye and nasal elevation and edema of the left optic nerve (Figure 6). Optic nerve head OCT of the right eye showed advanced, generalized thinning of both the GCC and RNFL (Figure 7). This correlated with the patient’s history of ON and her optic nerve presentation (pallor). Optic nerve head OCT scanning of the left eye revealed significant RNFL thickening combined with an inferior-temporal zone of GCC thinning (Figure 7). A recurrence of ON was suspected and repeat MRI of the brain and orbits was performed with and without contrast. The results were essentially normal with patchy, longitudinal enhancement of the left intraorbital segment of the optic nerve (Figure 8). AQP4-IgG and myelin oligodendrocyte glycoprotein (MOG) antibody testing was performed and showed positive anti-MOG titers. A diagnosis of MOGAD ON was established and the patient was treated with pulsed intravenous methylprednisolone followed by an oral prednisone taper. Follow-up visits showed rapid improvement of vision to 20/25 in the left eye in the presence of resultant disc pallor. No RAPD was noted. Repeat OCT showed stable findings of advanced and generalized thinning of both the GCC and RNFL in the right eye. OCT of the left eye showed advanced and generalized thinning of both the GCC and RNFL, which corresponded to the new disc pallor (Figure 9). In the next 2 years, the patient experienced several episodes of ON involving both the right and left eye. With each relapse, she reported rapid vision improvement following treatment with intravenous steroids. She was subsequently placed on maintenance therapy with oral MMF and her condition has remained stable with no further recurrences.
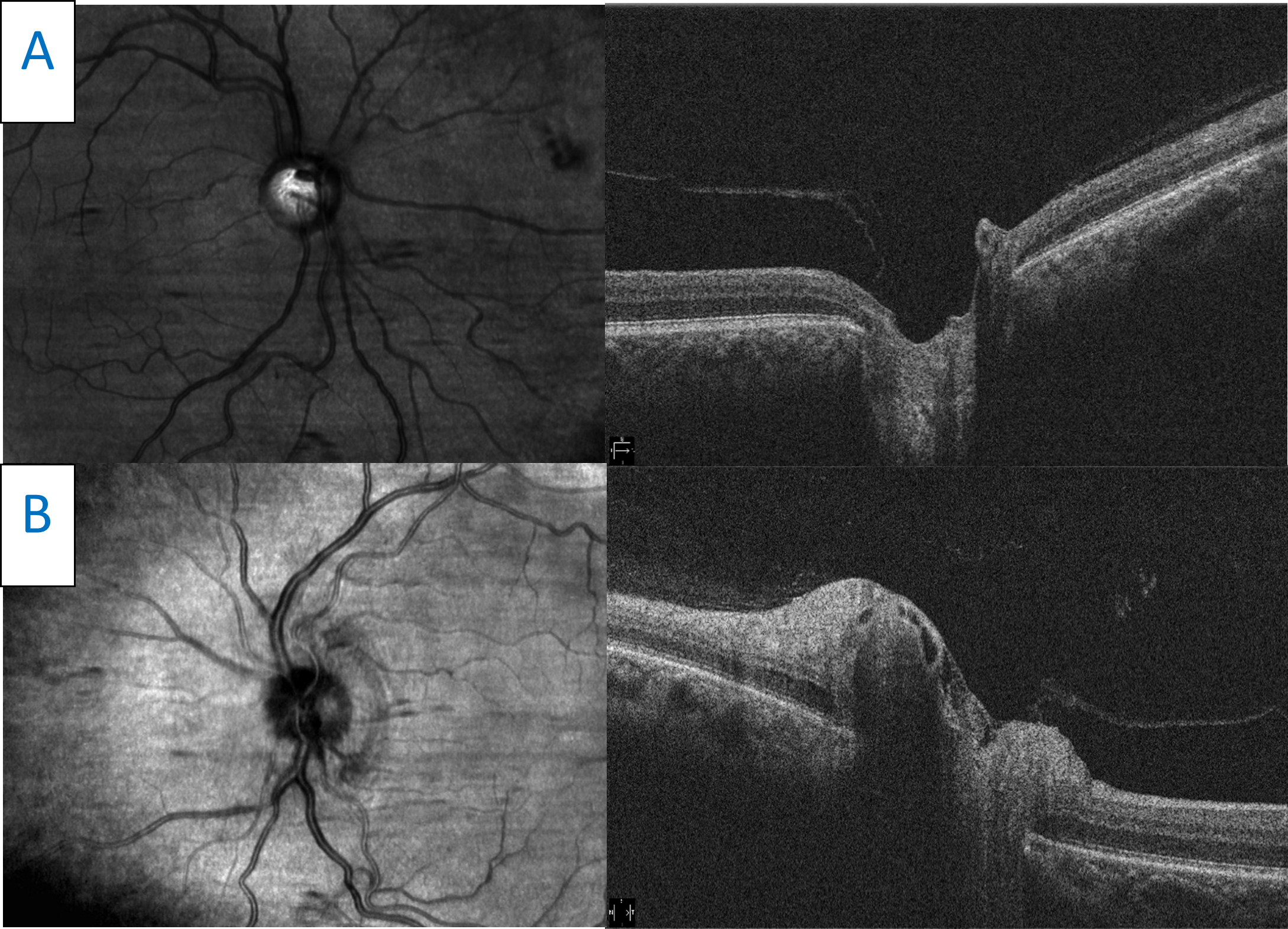 Figure 6. Horizontal raster scans taken through the center point of both optic nerves. A: The right optic disc is flat with significant cupping. B: The left optic disc scan shows moderate elevation and edema. Click to enlarge |
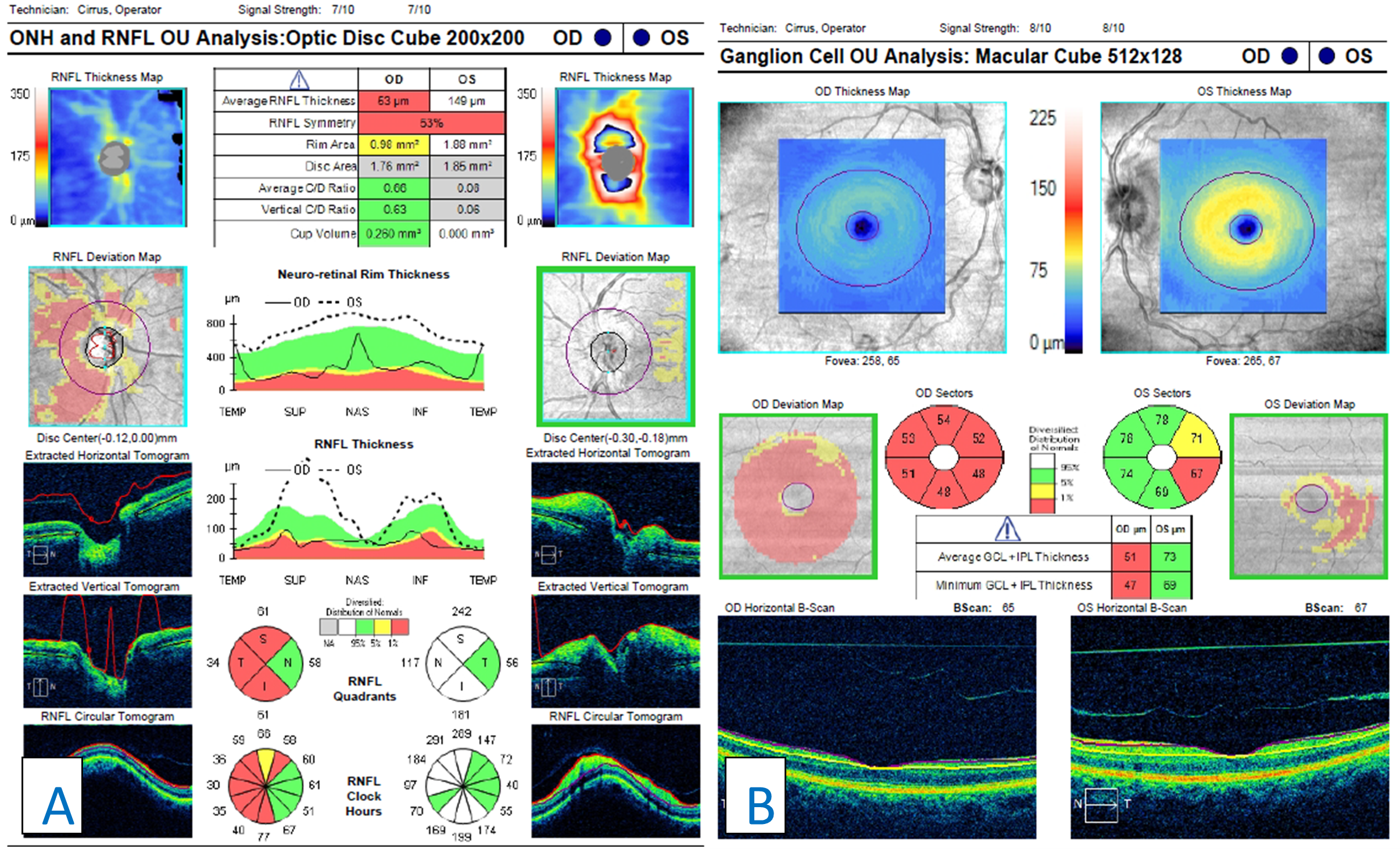 Figure 7. A: Retinal nerve fiber layer (RNFL) analysis of both eyes. In the right eye, advanced RNFL thinning, sparing the nasal portion of the nerve, is seen. Compared with the previous scans (Figure 5), progression is illustrated by a lower average thickness and deeper and larger amounts of thinning on the deviation map. Analysis of the left eye shows acute RNFL thickening, greater superiorly than inferiorly, which was not noted in the previous scans (Figure 5). B: Ganglion cell analysis of both eyes. Advanced ganglion cell complex (GCC) thinning is present in the right eye. This corresponds with the patient’s previous bout of optic neuritis. Compared with previous scans (Figure 5), progressive GCC thinning is illustrated in the right eye by a lower average GCC thickness. Inferior-temporal GCC thinning is noted in the left eye. The scan is relatively stable when compared with the previous scan (Figure 5). |
 Figure 8. A: T1-weighted coronal magnetic resonance imaging (MRI) with gadolinium showing mild enhancement of the left optic nerve (yellow arrow). B: T1-weighted axial MRI showing mild longitudinal enhancement of the left optic nerve (blue arrow). Click to enlarge |
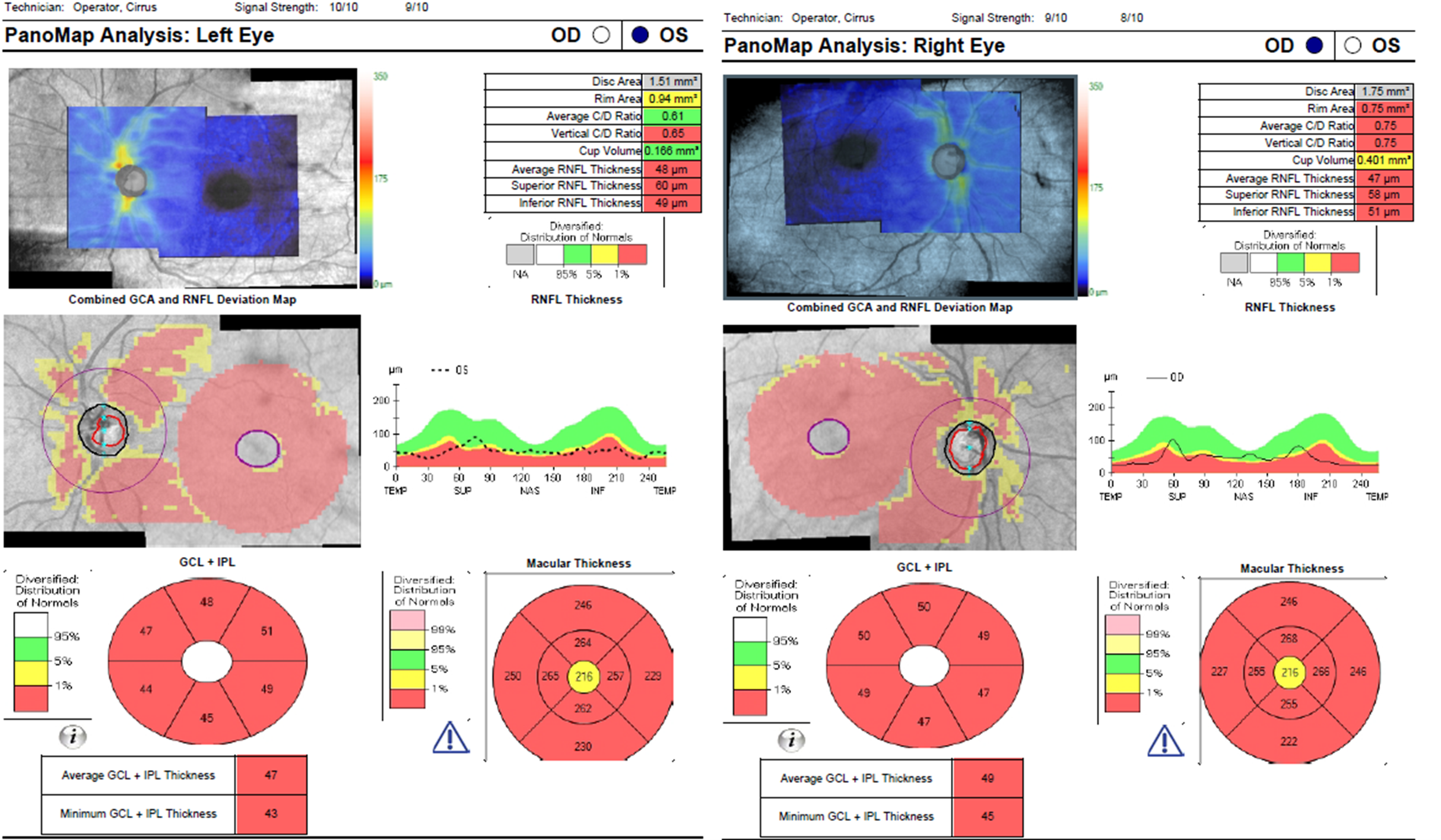 Figure 9. Panomap analysis showing advanced, bilateral thinning of the retinal nerve fiber layer and ganglion cell complex. These findings correspond to the bilateral disc pallor observed on fundus examination. Click to enlarge |
Education Guidelines
Key concepts
- The pathophysiology of MOGAD ON and NMOSD ON
- Clinical signs and symptoms to help differentiate between NMOSD ON, MOGAD ON and similar conditions
- Understanding medical treatments for both conditions
- Long-term vision implications for patients with either NMOSD ON or MOGAD ON
Learning objectives
- Define and recognize the clinical presentation of NMOSD ON and MOGAD ON, including signs and symptoms
- Know the importance of signs and symptoms and results of additional testing in determining a correct diagnosis
- Understand the pharmacologic treatments used for each condition
Discussion questions
- Knowledge, understanding and facts related to the case and condition
-
- Describe the typical appearance and presentation of NMOSD ON
- Describe the typical appearance and presentation of MOGAD ON
- Explain the pathogenesis of each condition. How are they similar? How do they differ?
- Differential diagnosis
-
- What other condition(s) should be considered as differential diagnoses for NMOSD ON and MOGAD ON?
- How can a clinician differentiate between these similar conditions based on presentation (history, signs, symptoms)?
- Patient management and role of the optometrist
-
- What additional testing should be ordered for MOGAD ON and NMOSD ON?
- What are the similarities and differences of test results for the respective conditions?
- Critical-thinking concepts
-
- When should a patient start pharmacological treatment for either condition?
- What medications are indicated initially for each condition? Why?
- What type of treatment outcomes can be expected for NMOSD and MOGAD?
Assessment of learning objectives
As MOGAD ON and NMOSD ON are fairly novel and advanced topics, these teaching case reports are best-suited for third- and fourth-year students and residents who have already been taught the foundational knowledge associated with ON, specifically MS-related ON. Formal assessment could be conducted in a variety of ways, such as:
- The case reports could be used as a part of a journal club at a student’s school, optometric rotation site or residency site. The students and/or residents could work in small groups or independently to answer the discussion questions. Open dialogue of the discussion questions would be encouraged so that the students and residents can learn from each other and from any mistakes made during the process. Follow-up meetings to discuss other specific literature cited in these case reports could also be scheduled.
- The cases could be presented to third- or fourth-year optometry students in a classroom setting. The case details could be presented to students who would then be responsible for arriving at a final diagnosis and proper management plan based on the given findings and test results. Comprehension and knowledge could be assessed through open-ended questioning to the class or through formal testing with multiple-choice questions.
- The case descriptions along with the results of any additional testing could be presented online to third- or fourth-year optometry students. Students could then be tested with multiple-choice questions on concepts involving pathophysiology, signs and symptoms, differential diagnosis, test interpretation, and treatment and management. Students would be required to answer a question correctly before moving on to the next question. Feedback and further elaboration on tested concepts could be provided if the question is answered correctly or incorrectly. This would allow students to learn from their potential mistakes and/or strengthen their existing knowledge base.
Discussion
While MS has been associated with ON in 57-80% of cases, clinicians must keep in mind that other etiologies exist. Studies have shown that idiopathic (14-29%), MOGAD (5-12%) and NMOSD (3-5%) ON can also occur.7-9 Clinical presentations of ON secondary to MS, NMOSD and MOGAD can be difficult to distinguish based on clinical findings and presentation alone.1,3,10 Typical MS ON presents as a demyelinating autoimmune condition at approximately 30 years of age. As the condition is autoimmune in origin, women are more likely to be affected. Patients usually present with unilateral visual acuity and visual field loss, a corresponding RAPD, dyschromatopsia and pain upon eye movement.1 The amount of visual acuity loss varies but has been found to be better than 20/200 in more than 50% of patients.1 Examination of the posterior segment can reveal disc swelling, or as seen in a majority of cases, the inflammation can be retrobulbar.11 Neuroimaging with MRI produces abnormal results with the presence of periventricular white matter lesions. In terms of treatment, in the Optic Neuritis Treatment Trial (ONTT) intravenous methylprednisolone led to faster vision recovery but had no real effect on the final visual outcome. ONTT also established that intravenous methylprednisolone treatment should be followed by oral prednisone as this led to lower rates of MS within the first 2 years of the ON attack.12
As not every presentation follows these norms or responds to the above treatment guidelines, cases outside these parameters should be considered atypical and etiologies such as NMOSD ON and MOGAD ON should be considered. Quick and accurate identification of underlying associated disorders in atypical acute ON episodes is important to prevent permanent vision loss.13 The risk of permanent vision loss ≤ 20/200 is approximately 3% for MS ON, 6-14% for MOGAD ON and > 33% for NMOSD ON. This underscores the importance of identifying non-MS causes of ON in a timely manner.1,10,14
NMOSD and MOGAD pathophysiology
The pathophysiology of NMOSD has been shown to be an autoimmune attack on AQP4 water channels in astrocyte foot processes (AQP4 immunoglobulin) in the central nervous system.1 The specific antibody that targets the AQP4 water channel on astrocytes is therefore referred to “AQP4-IgG” or “NMO-IgG.”1,2,15 This leads to complement activation and ultimately to secondary cytotoxic demyelination.1 Areas of the brain that are rich in AQP4 channels include the optic chiasm, spinal cord and the area postrema in the dorsal medulla. Thus, these areas are most commonly involved with NMOSD-associated disease.1 Discussion of the clinical phenotypes of NMOSD other than ON are beyond the scope of this paper but include transverse myelitis, area postrema syndrome (resultant nausea and vomiting), acute brainstem syndrome, narcolepsy and cerebral syndrome.1
The pathophysiology of MOGAD has also been shown to be an autoimmune attack; however, the pathophysiology differs when compared with NMOSD. MOG is a transmembrane protein expressed on myelin sheaths (MOG immunoglobulin) in the central nervous system.1,6,14 Antibodies targeting them lead to primary demyelination in the central nervous system, but notably spare astrocytes, unlike NMOSD.1,6,14 The clinical phenotype is also diverse when compared with NMOSD phenotype, but similarities exist as both conditions can present with ON and transverse myelitis. Interestingly, MOG-IgG is positive in approximately one-third of seronegative NMO patients notably in the setting of transverse myelitis.1,6,14 However, the diseases rarely co-exist due to their different mechanisms. Other signs seen with MOGAD include acute demyelinating encephalomyelitis and brainstem encephalitis.
Clinical presentation of NMOSD ON and MOGAD ON and comparisons to MS ON
Although NMOSD ON, MOGAD ON and MS ON all lead to demyelination, clinicians can use clinical presentation, neuroimaging and laboratory studies to help differentiate them. MOGAD ON has an equal prevalence among males and females with most patients initially presenting in their 30s.1,16 In contrast, NMOSD ON patients are much more likely to be female and to present in their 40s while MS ON patients are more likely to be females in their 30s.1 Clinicians should note that MOGAD ON can be present in childhood, which is rare with NMOSD.1,17 All three conditions may present with eye pain upon movement (of the involved eye), but pain is seen more frequently with MOGAD ON. Other characteristics include bilateral presentation (although MOGAD ON can be unilateral at times), severe vision loss, and recurrences for NMOSD ON and MOGAD ON.1 In contrast, MS ON presents unilaterally, thus it presents with a corresponding RAPD. It also has a possible relapsing course with the amount of vision loss at nadir having been found to be less severe when compared with NMOSD ON and MOGAD ON.1 Examination of the optic nerve reveals edema, which tends to be more severe in MOGAD ON than in NMOSD ON.1,17,18 MS ON is distinct in this sense as it tends to present as retrobulbar in a majority of cases. Therefore, no edema is visibile.11 In terms of outcomes, MOGAD ON patients tend to recover rapidly when intravenous steroids are initiated, which is not seen with NMOSD ON. As the recovery is rapid, MOGAD ON patients also tend to have better visual outcomes. Studies have indicated that only 6-10% of patients with MOGAD ON have final visual acuities worse than 20/200 compared to a third of NMOSD ON patients.1,16,19 Table 1 summarizes the similarities and differences between MOGAD ON, NMOSD ON and MS ON.
Neuroimaging
MRI findings between the conditions can be similar but with key differences. Enhancement of the ON with longer segments of involvement is seen with MOGAD ON and NMOSD ON, while MS ON tends to show shorter segments of involvement. MOGAD ON also shows enhancement of the perineural tissue of the optic nerve, which can also extend into the orbit. In contrast, NMOSD ON shows enhancement of the optic nerve extending to the optic chiasm and optic pathways.1 Thalamic and pontine lesions are more common in MOGAD ON than in NMOSD ON. MS-related brain neuroimaging is abnormal with the presence of periventricular white matter lesions not seen with either of the other two conditions. Cerebrospinal fluid analysis frequently shows oligoclonal bands that are rarely associated with the other two conditions.11
Laboratory testing
Serum samples are preferred over cerebrospinal fluid testing and should include AQP4-IgG cell assays as well as MOGAD IgG. The AQP4-IgG antibody has 75% sensitivity and > 99% specificity for NMOSD.1,15 In both cases presented, AQP4-IgG antibody and MOGAD IgG tests were ordered. The AQP4-IgG results were positive for the first patient, confirming NMOSD ON. MOGAD ON was confirmed for the second patient based on her positive MOGAD IgG titer.
Testing is more sensitive prior to initiation of treatment, and repeat testing can be considered a few months after an initial negative test if clinical signs and symptoms are strong for either condition. As ON can have many causes other than demyelinating conditions, clinicians should also be thorough in testing for infectious and inflammatory disease, such as syphilis and sarcoidosis.
Treatment and outcomes/prognosis
With phenotypic overlap, namely ON, it can be difficult to distinguish between NMOSD ON and MOGAD ON. Therefore, clinicians should be cognizant of the underlying etiology and results of ancillary testing. This can help guide decision-making toward the ideal treatment options and best possible outcomes.
MOGAD ON
Although standard treatment criteria and guidelines have not been created, general guidelines can be followed. The treatment of MOGAD ON can be grouped into acute and chronic options with overlap between the two. For acute bouts, and because 50% of MOGAD ON is monophasic, standard treatment is a 3- to 5-day course of intravenous high-dose steroid therapy, usually methylprednisolone.20 MOGAD ON responds more positively to steroids than MS ON and NMOSD ON do; however, high doses are indicated as lower doses have been associated with relapse.21 In one study, 95% of patients who were given at least 20 mg of prednisone for 6 months after the initial event had no repeat attacks for more than a year.21 After the initial high dose of intravenous steroids, oral prednisone can be initiated and then tapered for 1 to 3 months.1 Plasma exchange and intravenous immunoglobins are also options but should be reserved for cases of severe vision loss (< 20/100) or when recovery is not seen with initial intravenous steroid treatment.
As recovery occurs rapidly with positive vision outcomes with steroid treatment, long-term or chronic therapy is less likely to be needed. Clinicians should note, however, that even in the absence of corticosteroid treatment, many patients recover normal or near-normal visual acuity.22 Long-term maintenance therapy for single-event MOGAD ON is not indicated as 50% of these individuals remain monophasic. However, cases can be relapsing despite steroid treatment, and immunotherapy can be initiated to prevent long-term visual disability. Immunomodulators can also be used if the initial attack leads to residual deficits or if MOGAD-IgG titer is still positive 6 months after the initial attack, which have been associated with higher rates of relapse.16 Immunosuppressants that can be considered include rituximab, azathioprine (AZA) and MMF. This treatment paradigm was evident in our case #2. Although the patient initially responded well to steroid treatment, recurrences were noted necessitating chronic immunotherapy with MMF. Clinicians can also combine treatments to prevent further relapse as studies have shown that patients using oral steroids for an extended period in conjunction with immunosuppressive drugs were less likely to have a recurrent attack than patients taking only immunomodulators.19,21 Caution should be taken when prescribing rituximab because relapses have occurred after the first rituximab infusion in approximately 30% of patients.23 Promising results with newer monoclonal antibody treatments such as tocilizumab are emerging, but more research is required at this time.24,25 It must be remembered that these are off-label uses of these medications, and further clinical trials are necessary to confirm their therapeutic value.
NMOSD ON
Treatment for NMOSD ON can be similar to treatment for MOGAD; however, outcomes can be drastically different. As mentioned previously, despite treatment, NMOSD ON patients tend to have worse vision outcomes. Again, the treatment can be divided into acute and chronic categories with overlap between the two in certain situations. Treatment is further divided into classic or new treatment due to the introduction of novel medications. Although a multitude of treatment options exist, there are no clinically established guidelines, and treatment patterns vary from clinician to clinician.
Classic options for initial treatment of acute NMOSD ON include intravenous methylprednisolone, which is transitioned to a slow taper with oral steroids. Complete vision recovery is possible and is more likely when steroids are started promptly, underscoring the need for immediate treatment.20,26,27 Next, evidence exists for the benefit of plasma exchange, especially if a patient’s vision does not respond to initial steroid treatment. Similar to steroid treatment, plasma exchange should occur promptly as better outcomes have been noted when compared with delayed treatment.28 Plasma exchange can also be combined with steroid treatment, which can lead to better outcomes than with intravenous steroids alone.26 Next, although rarely given, due to limited data sets, immunoglobin therapy can also be considered.29,30 Newer options for acute disease are becoming widely available and include monoclonal antibody medications such as intravenous bevacizumab and ublituximab in conjunction with traditional intravenous steroid treatment. Although these medications show promise, data is limited at this time.31,32
Long-term or chronic therapy is prudent for patients with NMOSD as relapses are common and can lead to further disability. Again, with recent advances, therapy can now be classified as classic or new.33
Classic therapy for chronic NMOSD consists of immunosuppressive agents such as AZA, MMF, rituximab and tocilizumab. Numerous studies have shown AZA and MMF to be effective in terms of preventing relapses.18,33-36 When comparing the two drugs, MMF proved to be superior in terms of efficacy in preventing annual relapses and in terms of side effects.37 The effects of this treatment approach were noted in our case #1. The patient was initially started on systemic steroids as was warranted. He was then transitioned to chronic treatment with MMF with no recurrence to date. Unfortunately, these medications usually take 4 to 6 months to exert their clinical effect and therefore must be given in conjunction with oral steroids.33
Ritixumab is another option for clinicians. It has been shown to be more effective than AZA or MMF in terms of relapse severity and relapse prevention.33,38-40 Although the number of randomized controlled studies is limited, one multicenter randomized double-blind placebo-controlled study by Tahara et al. revealed that 7 of 9 patients treated with placebo relapsed vs. none treated with rituximab.40 It can also be considered when there are contraindications to MMF and AZA. The monoclonal antibody tocilizumab can also be considered. It has been shown to be effective in stabilizing relapses when compared with the previously mentioned treatment options and can be used as an alternative for patients who do not respond appropriately to ritixumab.33,41,42
With advancements in the understanding of the pathophysiology of NMOSD, three newer monoclonal antibody treatments have been approved by the FDA: eclulizamab, inebilizumab and satralizumab.33 Each offers a distinctive mechanism of action in terms of preventing NMOSD relapses.
Eculizumab targets the terminal complement system to decrease inflammation.43 Although previous studies showed eculizumab to be beneficial,44 the seminal study was the PREVENT trial, which assessed the efficacy and safety of eculizumab as add-on or monotherapy compared with placebo. Results showed a 94% reduction in relapse risk in patients with NMOSD.44 Inebilizumab decreases NMOSD relapses by depleting CD19-positive B-cells.18,45 The largest study in NMOSD to date was the N-MOmentum trial, which showed a 73% relative risk reduction in number of relapses with inebilizumab vs. placebo. A more robust response was noted in patients who were positive for the AQP4 antibody.45 Satralizumab targets the interleukin 6 (IL-6) receptor.17 The cytokine IL-6 is thought to be a key driver of inflammation in NMOSD. The phase 3 SAkuraSky study showed a 62% reduction in relapse risk when satralizumab was added to immunosuppressive treatment.46
Although many treatments exist for NMOSD, most are based on small and retrospective studies. New treatment options show promise; however, long-term data on them is not available. Ultimately, large multicenter studies for a longer period of time are needed to establish clinical guidelines.
Conclusion
The historical paradigm of ON management is evolving. In our opinion, standard of care for ON should now include testing for NMOSD and MOGAD biomarkers in all acute cases. This allows earlier identification and potentially better outcomes in NMOSD- and MOGAD-associated ON, which have worse vision prognoses compared with MS ON.1,10,14 Neuroimaging has previously been recommended for all cases of acute ON and should be standard of care going forward.1,3,47 Promising new treatment options for both conditions, which differ from treatments used for MS ON, are now available. Therefore, it is appropriate to recommend utilizing MOG-IgG and NMO-IgG testing/titers for all cases of acute ON in hopes of achieving optimal outcomes.
References
- Chen JJ, Pittock SJ, Flanagan EP, Lennon VA, Bhatti MT. Optic neuritis in the era of biomarkers. Surv Ophthalmol. 2020 Jan-Feb;65(1):12-17. doi: 10.1016/j.survophthal.2019.08.001.
- Gospe SM 3rd, Chen JJ, Bhatti MT. Neuromyelitis optica spectrum disorder and myelin oligodendrocyte glycoprotein associated disorder-optic neuritis: a comprehensive review of diagnosis and treatment. Eye (Lond). 2021 Mar;35(3):753-768. doi: 10.1038/s41433-020-01334-8.
- De Lott LB, Bennett JL, Costello F. The changing landscape of optic neuritis: a narrative review. J Neurol. 2022 Jan;269(1):111-124. doi: 10.1007/s00415-020-10352-1.
- Fujihara K, Cook LJ. Neuromyelitis optica spectrum disorders and myelin oligodendrocyte glycoprotein antibody-associated disease: current topics. Curr Opin Neurol. 2020 Jun;33(3):300-308. doi: 10.1097/WCO.0000000000000828.
- Seay M, Rucker JC. Neuromyelitis optica: review and utility of testing aquaporin-4 antibody in typical optic neuritis. Asia Pac J Ophthalmol (Phila). 2018 Jul-Aug;7(4):229-234. doi: 10.22608/APO.2018170.
- Chen JJ, Bhatti MT. Clinical phenotype, radiological features, and treatment of myelin oligodendrocyte glycoprotein-immunoglobulin G (MOG-IgG) optic neuritis. Curr Opin Neurol. 2020 Feb;33(1):47-54. doi: 10.1097/WCO.0000000000000766.
- Hassan MB, Stern C, Flanagan EP, et al. Population-based incidence of optic neuritis in the era of aquaporin-4 and myelin oligodendrocyte glycoprotein antibodies. Am J Ophthalmol. 2020 Dec;220:110-114. doi: 10.1016/j.ajo.2020.07.014.
- Deschamps R, Lecler A, Lamirel C, et al. Etiologies of acute demyelinating optic neuritis: an observational study of 110 patients. Eur J Neurol. 2017 Jun;24(6):875-879. doi: 10.1111/ene.13315.
- Etemadifar M, Abbasi M, Salari M, Etemadifar F, Tavakoli H. Comparing myelin oligodendrocyte glycoprotein antibody (MOG-Ab) and non MOG-Ab associated optic neuritis: Clinical course and treatment outcome. Mult Scler Relat Disord. Jan 2019;27:127-130. doi:10.1016/j.msard.2018.10.013.
- Beck RW, Gal RL, Bhatti MT, et al.; Optic Neuritis Study Group. Visual function more than 10 years after optic neuritis: experience of the optic neuritis treatment trial. Am J Ophthalmol. 2004 Jan;137(1):77-83. doi: 10.1016/s0002-9394(03)00862-6. Erratum in: Am J Ophthalmol. 2004 Apr;137(4):following 793. Erratum in: Am J Ophthalmol. 2004 Aug;138(2):following 321.
- Wynford-Thomas R, Jacob A, Tomassini V. Neurological update: MOG antibody disease. J Neurol. May 2019;266(5):1280-1286. doi:10.1007/s00415-018-9122-2.
- Beck RW, Gal RL. Treatment of acute optic neuritis: a summary of findings from the optic neuritis treatment trial. Arch Ophthalmol. 2008 Jul;126(7):994-5. doi: 10.1001/archopht.126.7.994.
- Jenkins TM, Toosy AT. Optic neuritis: the eye as a window to the brain. Curr Opin Neurol. 2017 Feb;30(1):61-66. doi: 10.1097/WCO.0000000000000414.
- Kaushik M, Burdon MA. Myelin oligodendrocyte glycoprotein antibody-associated optic neuritis – a review. J Neuroophthalmol. 2021 Dec 1;41(4):e786-e795. doi: 10.1097/WNO.0000000000001234.
- Wu Y, Zhong L, Geng J. Neuromyelitis optica spectrum disorder: pathogenesis, treatment, and experimental models. Mult Scler Relat Disord. Jan 2019;27:412-418. doi:10.1016/j.msard.2018.12.002.
- Jurynczyk M, Messina S, Woodhall MR, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017 Dec 1;140(12):3128-3138. doi: 10.1093/brain/awx276. Erratum in: Brain. 2018 Apr 1;141(4):e31.
- Carnero Contentti E, Correale J. Neuromyelitis optica spectrum disorders: from pathophysiology to therapeutic strategies. J Neuroinflammation. 2021 Sep 16;18(1):208. doi: 10.1186/s12974-021-02249-1.
- Held F, Klein AK, Berthele A. Drug treatment of Neuromyelitis optica spectrum disorders: out with the old, in with the new? Immunotargets Ther. 2021 Mar 19;10:87-101. doi: 10.2147/ITT.S287652.
- Jarius S, Ruprecht K, Kleiter I, et al.; in cooperation with the Neuromyelitis Optica Study Group (NEMOS). MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation. 2016 Sep 27;13(1):280. doi: 10.1186/s12974-016-0718-0.
- Stiebel-Kalish H, Hellmann MA, Mimouni M, Paul F, Bialer O, Bach M, Lotan I. Does time equal vision in the acute treatment of a cohort of AQP4 and MOG optic neuritis? Neurol Neuroimmunol Neuroinflamm. 2019 May 21;6(4):e572. doi: 10.1212/NXI.0000000000000572.
- Ramanathan S, Mohammad S, Tantsis E, et al.; Australasian and New Zealand MOG Study Group. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. 2018 Feb;89(2):127-137. doi: 10.1136/jnnp-2017-316880.
- Chen JJ, Tobin WO, Majed M, et al. Prevalence of myelin oligodendrocyte glycoprotein and aquaporin-4-IgG in patients in the Optic Neuritis Treatment Trial. JAMA Ophthalmol. 2018 Apr 1;136(4):419-422. doi: 10.1001/jamaophthalmol.2017.6757.
- Durozard P, Rico A, Boutiere C, et al. Comparison of the response to rituximab between myelin oligodendrocyte glycoprotein and aquaporin-4 antibody diseases. Ann Neurol. 2020 Feb;87(2):256-266. doi: 10.1002/ana.25648.
- Elsbernd P, Hoffmann W, Wingerchuk D, Carter J. Interleukin-6 inhibition with tocilizumab for relapsing MOG-IgG associated disorder (11). Neurology. April 2021;96 (15 Supplement) 11.
- Lu Q, Luo J, Hao H, et al. Efficacy and safety of long-term immunotherapy in adult patients with MOG antibody disease: a systematic analysis. J Neurol. Dec 2021;268(12):4537-4548. doi:10.1007/s00415-020-10236-4.
- Abboud H, Petrak A, Mealy M, Sasidharan S, Siddique L, Levy M. Treatment of acute relapses in neuromyelitis optica: steroids alone versus steroids plus plasma exchange. Mult Scler. Feb 2016;22(2):185-92. doi:10.1177/1352458515581438.
- Kleiter I, Gahlen A, Borisow N, et al. Neuromyelitis optica: evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. Feb 2016;79(2):206-16. doi:10.1002/ana.24554.
- Bonnan M, Valentino R, Debeugny S, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. Apr 2018;89(4):346-351. doi:10.1136/jnnp-2017-316286.
- Elsone L, Panicker J, Mutch K, Boggild M, Appleton R, Jacob A. Role of intravenous immunoglobulin in the treatment of acute relapses of neuromyelitis optica: experience in 10 patients. Mult Scler. Apr 2014;20(4):501-4. doi:10.1177/1352458513495938.
- Li X, Tian DC, Fan M, et al. Intravenous immunoglobulin for acute attacks in neuromyelitis optica spectrum disorders (NMOSD). Mult Scler Relat Disord. 2020 Sep;44:102325. doi: 10.1016/j.msard.2020.102325.
- Mealy MA, Levy M. A pilot safety study of ublituximab, a monoclonal antibody against CD20, in acute relapses of neuromyelitis optica spectrum disorder. Medicine (Baltimore). 2019 Jun;98(25):e15944. doi: 10.1097/MD.0000000000015944.
- Mealy MA, Shin K, John G, Levy M. Bevacizumab is safe in acute relapses of neuromyelitis optica. Clin Exp Neuroimmunol. 2015 Nov 1;6(4):413-418. doi: 10.1111/cen3.12239.
- Carnero Contentti E, Marrodan M, Correale J. Emerging drugs for the treatment of adult MOG-IgG-associated diseases. Expert Opin Emerg Drugs. 2021 Jun;26(2):75-78. doi: 10.1080/14728214.2021.1919082.
- Bichuetti DB, Perin MMM, Souza NA, Oliveira EML. Treating neuromyelitis optica with azathioprine: 20-year clinical practice. Mult Scler. Jul 2019;25(8):1150-1161. doi:10.1177/1352458518776584.
- Nikoo Z, Badihian S, Shaygannejad V, Asgari N, Ashtari F. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol. Sep 2017;264(9):2003-2009. doi:10.1007/s00415-017-8590-0.
- Montcuquet A, Collongues N, Papeix C, et al. Effectiveness of mycophenolate mofetil as first-line therapy in AQP4-IgG, MOG-IgG, and seronegative neuromyelitis optica spectrum disorders. Mult Scler. Sep 2017;23(10):1377-1384. doi:10.1177/1352458516678474.
- Yang Y, Wang CJ, Wang BJ, Zeng ZL, Guo SG. Comparison of efficacy and tolerability of azathioprine, mycophenolate mofetil, and lower dosages of rituximab among patients with neuromyelitis optica spectrum disorder. J Neurol Sci. 2018 Feb 15;385:192-197. doi: 10.1016/j.jns.2017.12.034.
- Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol. Mar 2014;71(3):324-30. doi:10.1001/jamaneurol.2013.5699.
- Stellmann JP, Krumbholz M, Friede T, et al. Immunotherapies in neuromyelitis optica spectrum disorder: efficacy and predictors of response. J Neurol Neurosurg Psychiatry. Aug 2017;88(8):639-647. doi:10.1136/jnnp-2017-315603.
- Tahara M, Oeda T, Okada K, et al. Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. Apr 2020;19(4):298-306. doi:10.1016/s1474-4422(20)30066-1.
- Araki M, Matsuoka T, Miyamoto K, et al. Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology. Apr 15 2014;82(15):1302-6. doi:10.1212/wnl.0000000000000317.
- Zhang C, Zhang M, Qiu W, et al.; TANGO Study Investigators. Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (TANGO): an open-label, multicentre, randomised, phase 2 trial. Lancet Neurol. 2020 May;19(5):391-401. doi: 10.1016/S1474-4422(20)30070-3.
- Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. Jun 2013;12(6):554-62. doi:10.1016/s1474-4422(13)70076-0.
- Pittock SJ, Berthele A, Fujihara K, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med. 2019 Aug 15;381(7):614-625. doi: 10.1056/NEJMoa1900866.
- Cree BAC, Bennett JL, Kim HJ, et al.; N-MOmentum study investigators. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019 Oct 12;394(10206):1352-1363. doi: 10.1016/S0140-6736(19)31817-3.
- Yamamura T, Kleiter I, Fujihara K, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. 2019 Nov 28;381(22):2114-2124. doi: 10.1056/NEJMoa1901747.
- Traboulsee A, Simon JH, Stone L, et al. Revised recommendations of the Consortium of MS Centers Task Force for a standardized MRI protocol and clinical guidelines for the diagnosis and follow-up of multiple sclerosis. AJNR Am J Neuroradiol. 2016 Mar;37(3):394-401. doi: 10.3174/ajnr.A4539.

