
PEER REVIEWED
Granular Corneal Dystrophy
Raman Bhakhri, OD, FAAO, Kathryn Hohs, OD, FAAO, Chandler Wolfe, Aleeza Tariq
Abstract
Granular corneal dystrophy is an inherited corneal stromal dystrophy secondary to genetic mutations in the TGFBI gene. This mutation causes irregularities in the keratoepithelin protein leading to corneal stromal deposits. The condition is further divided into two types of granular corneal dystrophy, type 1 and type 2. These types have a similar phenotypic expression early in the disease process and therefore they may be indistinguishable.1 This case series highlights the utilization of genetic testing to confirm the ultimate diagnosis of granular corneal dystrophy 1 in a set of brothers. The pathophysiology, clinical findings, multimodal imaging results, as well as surgical and non-surgical management options for both dystrophies are also discussed.
Keywords
Introduction
Granular corneal dystrophy (GCD) is an autosomal dominant inherited corneal epithelial stromal dystrophy that results in the deposition of discrete and irregular white-grey deposits at the level of the corneal epithelium and anterior stroma.1,2 Different genetic mutations in the TGFBI (transforming growth factor beta induced) gene, specifically at the 5q31 locus, result in either of two subtypes of GCD, type 1 (GCD1) or type 2 (GCD2).3 Regardless of subtype, both conditions can potentially cause significant visual symptoms including loss of vision and photophobia secondary to progression and coalescing of deposits over time.4 The conditions can be diagnosed with careful slit lamp examination, in-vivo imaging, and a thorough history indicating potential involvement of other family members. However, definitive diagnosis is made through genetic testing, as at times the subtypes are indistinguishable.4,5 While no cure exists for either subtype, non-surgical and surgical treatments are available to patients.6,7 This case series presents two brothers who were ultimately diagnosed with GCD1. The pathophysiology, differential diagnoses, adjunct testing results and potential treatment options are discussed.
Patient 1 Case Report
A 23-year-old Hispanic male presented for an ocular health examination. He reported being diagnosed at age 16 with an unknown corneal dystrophy and was informed at that visit that no treatment was available. The patient reported no specific ocular complaints but wanted to learn more about his condition and any potential treatment options. His last eye exam was reported to be about 3 years ago at another office at which time a new prescription for glasses was given. However, the unknown corneal dystrophy was not further investigated. The patient denied any history of ocular surgeries or trauma. The patient’s medical history was unremarkable. He also denied any current or past medication use or any allergies. His family’s medical history was unremarkable; however, their ocular history was significant for an unknown corneal dystrophy for his mother, younger brother, maternal uncle, maternal grandmother and maternal cousin.
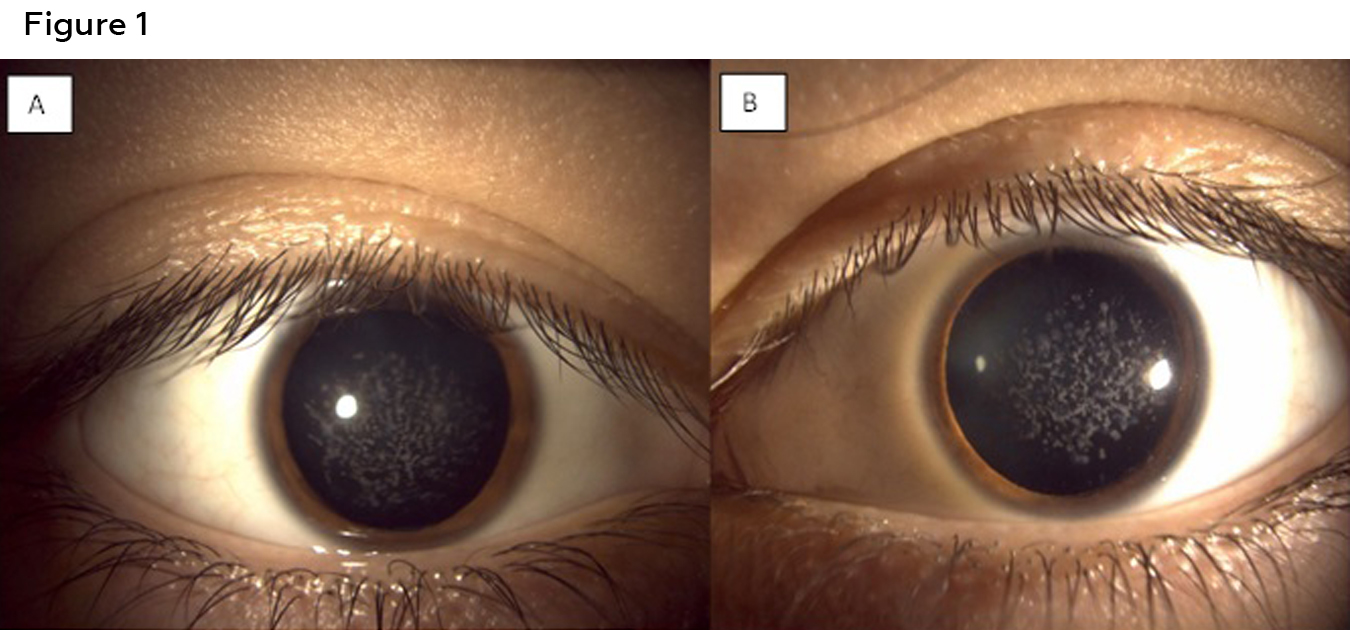
Figure 1: Anterior segment photos of the right (A) and left (B) eye of patient 1 showing white bread crumb like deposits that are concentrated more centrally and separated by clear corneal spaces. Click to enlarge
Best corrected visual acuities were 20/25 in the right eye and 20/30 in the left eye. Confrontation visual fields, pupils, and extra ocular motility testing were unremarkable. Biomicroscopic examination of the anterior segment revealed diffuse central breadcrumb-like deposits in the anterior stromal layers of both eyes. The deposits were irregular in shape with rough edges (Figure 1). The density and central location of the deposits likely accounted for the mildly reduced acuities. All other anterior segment structures were unremarkable. Intraocular pressures were 16mmHg in both eyes with Goldmann applanation tonometry. Dilated fundus examination was unremarkable in each eye. An anterior segment optical coherence tomography (OCT) (Zeiss, Dublin CA) scan of each eye was obtained. The OCT for each eye revealed hyper-reflective deposits in the anterior stroma of the cornea with clear intervening areas. Posterior shadowing was also seen in the stroma, correlating with hyper-reflective deposits seen anteriorly (Figure 2). Genetic testing was ordered (Avagen, Menlo Park, CA); results were positive for a heterozygous mutation in TGFBI, c.1663C>T, p.R555W., a mutation known to cause GCD1. The patient was informed of his diagnosis and was advised to contact the genetic counselor made available to him through the initial genetic testing to discuss the autosomal dominant inheritance pattern of the mutation. As his family history suggested that other family members likely have GCD1, he was advised to encourage these family members to present for examination as well. Unfortunately, all other family members were out-of-state, with the exception of a younger brother, who presented for examination (Case 2).
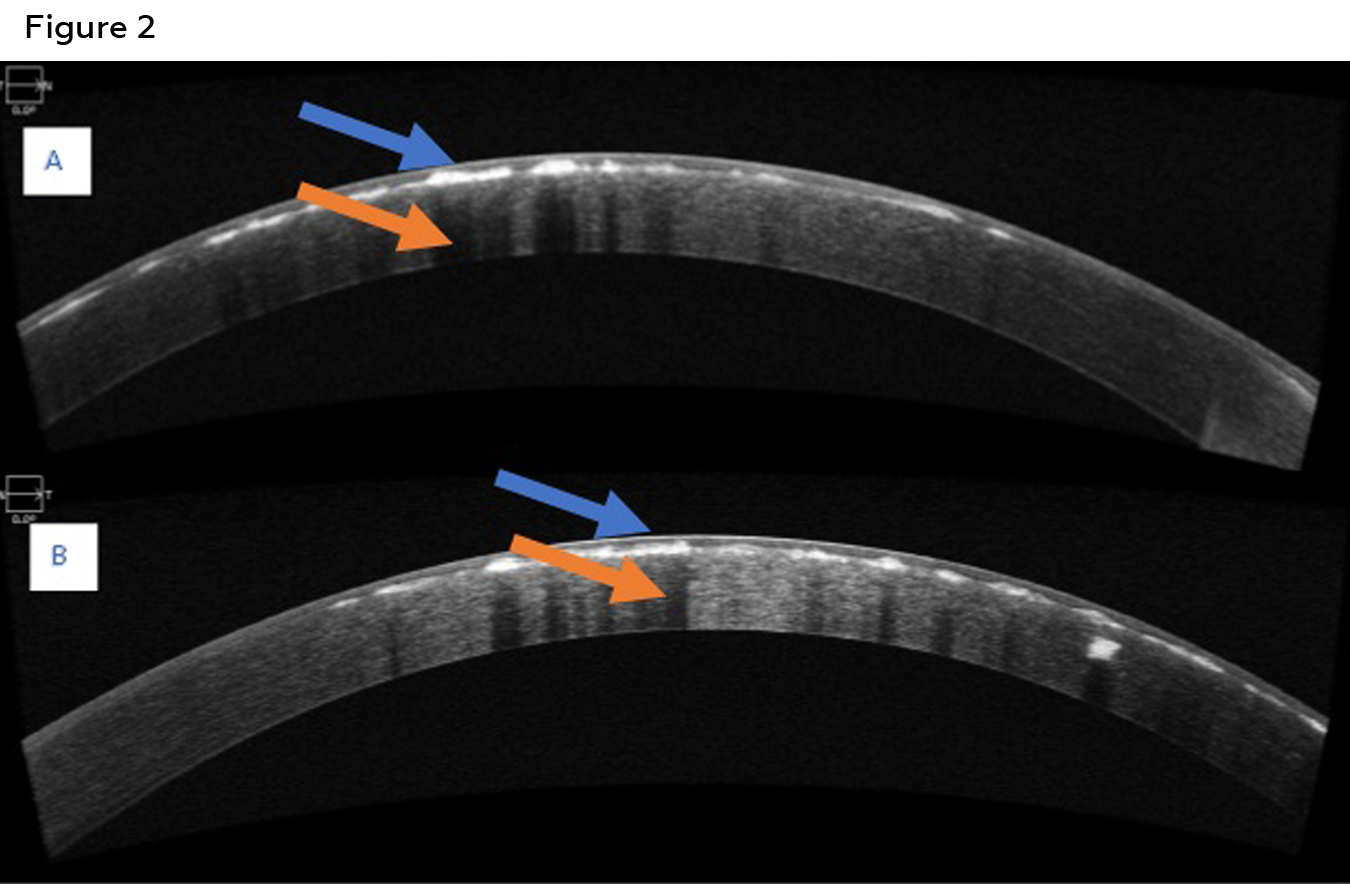
Figure 2: Anterior segment OCT of the right eye (A) and left eye (B) of patient 1 showing hyper reflective deposits at the level of anterior stroma (blue arrows). Posterior shadowing of the cornea secondary to the deposits is also visible (orange arrow). Click to enlarge
As the patient was asymptomatic, had no history of any prior instances of corneal erosion, and with the natural progression of the condition known, annual monitoring of the condition with repeat imaging was suggested to monitor for and document any objective evidence of progression. The patient was also informed that corneal surgery was not needed at this time based on the relatively stable and good visual acuities. The patient, however, requested a consultation for possible removal of the deposits. A referral was made per his request to a corneal specialist. The corneal specialist agreed with the initial recommendations and also advised annual monitoring.
Patient 2 Case Report
A 20-year-old Hispanic male presented with suspected GCD1 based on confirmation of GCD1 in his older brother. The patient reported constant blurry vision at distance and near in both eyes. No other ocular complaints were stated. The patient’s last eye exam was unknown and was described to be many years ago at a different location. He was given glasses at the visit but was not compliant in wearing them. The patient denied any history of ocular surgeries or trauma. The patient’s medical history was unremarkable. He also denied any current or past medication use or any allergies. His family’s medical history was unremarkable. Family ocular history was positive for confirmed GCD1 for his older brother and likely/but not confirmed GCD1 for his mother, maternal uncle, maternal grandmother and maternal cousin.
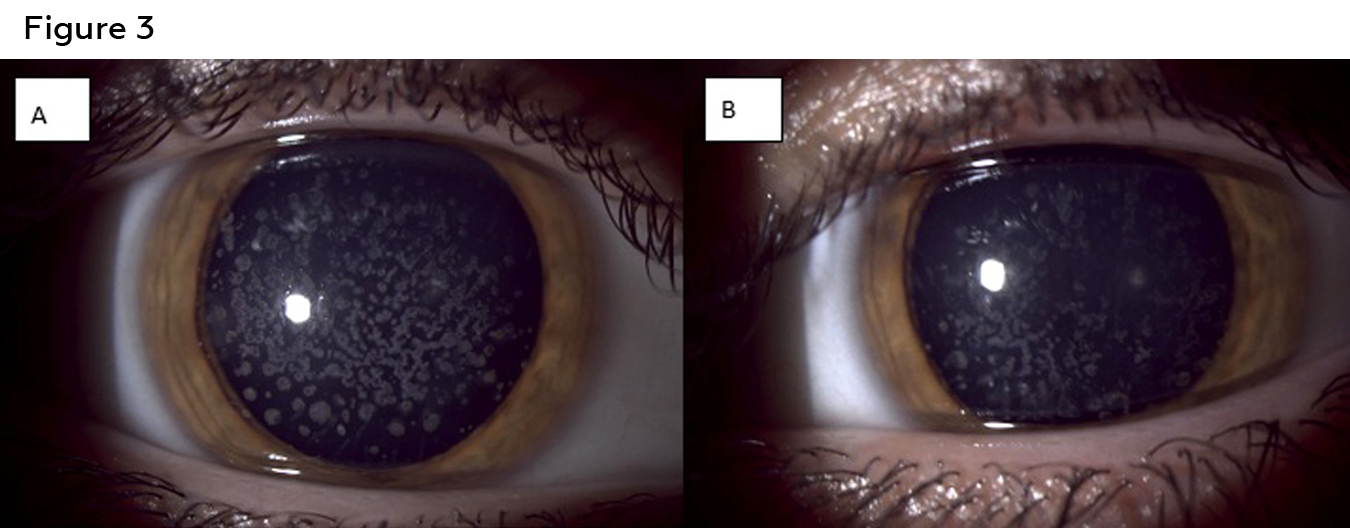
Figure 3: Anterior segment photos of the right (A) and left (B) eye of patient two showing white bread crumb like deposits that are concentrated more centrally and separated by clear corneal spaces. Click to enlarge
Best corrected visual acuity was 20/25 in the right and left eye. Confrontation visual fields, pupils and extra ocular motility testing were unremarkable. Intraocular pressures were 11mmHg in the right eye and 9mmHg in the left eye with Goldmann applanation tonometry. Examination of the anterior segment revealed diffuse central breadcrumb like deposits in the anterior stromal layers of both eyes. The deposits were irregular in shape with rough edges (Figure 3). The precipitates appeared irregular with rough edges and clear spaces between them. The density of the deposits centrally likely accounted for the mildly reduced acuity in each eye. All other anterior segment structures were unremarkable. Dilated fundus examination was also unremarkable in each eye. Anterior segment OCT showed similar findings to those of the patient’s older brother: hyper-reflective deposits in the anterior stroma of the cornea with corresponding posterior shadowing (Figure 4). Genetic testing was also ordered for the patient, with the results showing a heterozygous mutation similar to that of the older brother, TGFBI, c.1663C>T, p.R555W. As the patient was asymptomatic and had no history of any prior instances of corneal erosion, annual monitoring of the condition with repeat imaging was suggested. The patient was also informed that corneal surgery was not needed at this time based on the relative stability and good visual acuities.
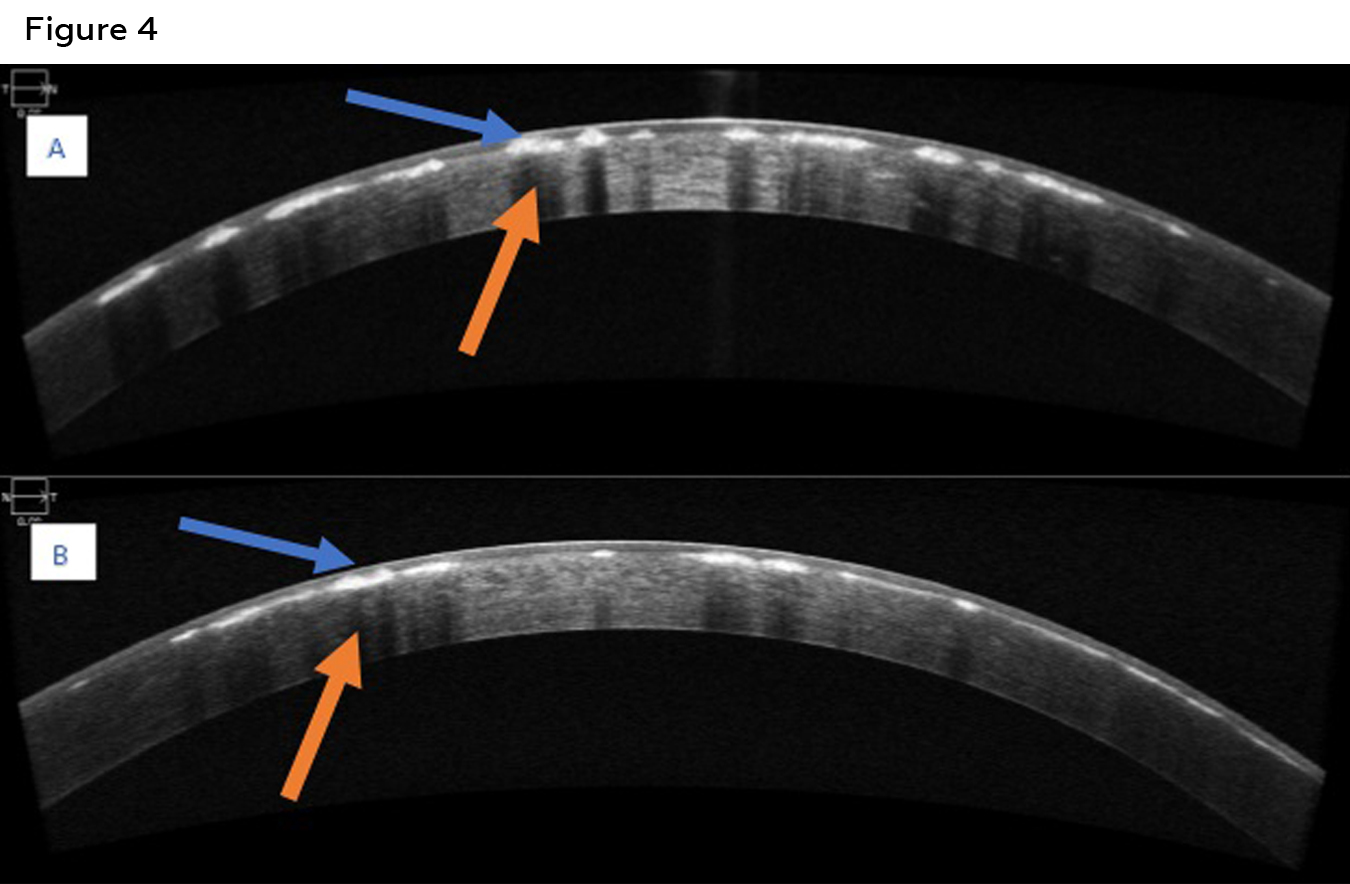
Figure 4: Anterior segment OCT of the right eye (A) and left eye (B) of patient two showing hyper reflective deposits at the level of anterior stroma (blue arrows). Posterior shadowing of the cornea secondary to the deposits is also visible (orange arrow). Click to enlarge
Educator’s Guide
Key Concepts
- The pathophysiology and specific genetics of GCD1 and GCD2
- The value of ancillary testing in GCD1 and GCD2
- Available treatment options for patients with GCD1 or GCD2
Learning Objectives
- Define and identify phenotypic variations in GCD1 and GCD2
- Compare and contrast other corneal epithelial stromal dystrophies as differential diagnoses for GCD1 and GCD2
- Understand and interpret in vivo imaging to assist in the diagnosis and management of GCD1 and GCD2
- Review existing non-surgical and surgical treatment options GCD1 and GCD2 and their outcomes
Discussion Questions
- Knowledge, understanding and facts about the clinical case and condition presentation
- Define the characteristic appearance of GCD1
- Define the characteristic appearance of GCD2
- Compare and contrast the genetics of these two conditions
- Differential diagnosis
- What other corneal dystrophies present with a phenotype similar to GCD1 and/or GCD2?
- How would a clinician distinguish between these conditions based on the patient’s phenotype, history (personal and family) and multimodal imaging results?
- Patient management and role of the optometrist
- What supplementary testing should be considered for patients and their family members who possibly have GCD1 or GCD 2?
- What surgical and non-surgical options are available and when are they indicated?
- How would you educate them on the prognosis of both conditions?
- Critical-thinking concepts
- Understand the many different presentations of TGFBI mutations and their role in corneal epithelial stromal dystrophies.
- How might reduced visual acuities and increased glare affect activities of daily living?
Assessment of Learning Objectives
GCD1 and GCD2 are corneal epithelial stromal dystrophies with many differential diagnoses because different mutations in a single gene, TGFBI, result in different disorders. Since a basic understanding of corneal anatomy, physiology, genetics and familiarity with multimodal imaging is needed for diagnosis and treatment, these cases are suitable for optometry students in their second year and beyond. The cases can also be utilized by residents and academic and non-academic providers. Assessments can include, but are not limited to, the following:
- Presenting the cases in a small or large group setting. Large group settings can include formal lectures on corneal dystrophies, in a student’s anterior segment/cornea class. These cases could comprise a part or section of the overall lecture(s) on corneal dystrophies or could be presented as a stand-alone lecture. Understanding and comprehension of the material could be achieved through answering the discussion questions in class or as an individual assignment to be completed and submitted at a later date. Immediate comprehension could be assessed through open-ended questions by the lecturer or through the use of testing technologies such as Turning Point. Formal understanding could be achieved through traditional means such as midterm and final examinations consisting of questions of the examiner’s choice (fill in the blank, multiple choice, essay, etc).
- A small group format could also be utilized, implemented in student labs for anterior segment and cornea, in which this format would be more feasible. Students could be presented initially with the case history and presentation. From there, students could discuss the case as a group and formulate an appropriate diagnosis with a corresponding treatment and management plan.
- A small group setting could also be used by residents in which the cases are discussed. Cited articles could also be used for further review.
- Lastly, independent reading of the article could be utilized by practicing and non-practicing clinicians who could answer the discussion questions while referencing the article.
Discussion
Corneal dystrophies represent a rare group of genetic conditions arising from mutations in various genes. These mutations result in corneal deposits with varying ages of onset, inheritance patterns (dominant, recessive or X-Linked), and clinical courses. At times, the dystrophies resemble each other making the diagnosis difficult and uncertain. In general, corneal dystrophies are classified based on the location of the deposits in the cornea. However, the most recent classification, referred to as the IC3D Classification of Corneal Dystrophies, considers traditional definitions together with the results of genetic testing and histology. Specifically, the dystrophies are classified as being either epithelial and subepithelial, epithelial-stromal TGFBI, stromal, or endothelial.1
GCD is an autosomal-dominant, inherited corneal condition characterized by the bilateral accumulation of distinct whitish deposits in the corneal stroma and epithelium not due to inflammation, trauma or systemic disease.1-5 Granular corneal dystrophy is considered an epithelial stromal dystrophy based on this classification and is further differentiated into two subtypes: GCD1 and GCD2.1,6
GCD1 occurs from mutations in the TGFBI gene which encodes for a keratoepithelin. This protein is thought to be responsible for corneal cell adhesion, movement and expansion.7 Mutations and impairments in this gene lead to the expression and production of abnormal hyaline deposits. The specific mutation seen is pArg555Trp, resulting in a phenotype that presents with multiple and small distinct whitish/gray granules that deposit in the anterior stroma.8 The phenotype can vary, as the deposition has also been observed as drops, crumbs or rings.9 Initially, clear areas of cornea can be visualized between the deposits with the condition being isolated to the central cornea. Patients at this early stage can be asymptomatic or can present with mild reductions in vision with accompanying glare. Visual acuity is not expected to improve with pinhole testing, as the reduction stems from stromal opacities rather than uncorrected refractive error or higher-order aberrations. As the condition progresses, the clear area between the granular deposits diminishes and the number of granules also increases, leading to further vision loss, glare and the potential for the development of recurrent corneal erosions (RCE).3,8,10 Corneal dystrophies, such as GCD, but namely epithelial basement membrane dystrophy, have been widely implicated in the development of RCEs. The mechanism is damage to the corneal hemi-desmosomes caused by the stromal deposits. These hemi-desmosomes play an important part in providing a link or anchor between the corneal epithelium, its basement membrane and deeper layers (Bowman’s layer and stroma).11 Homozygote cases tend to have more apparent and aggressive signs and symptoms compared to those experienced by heterozygotes.1,4,12 Both of our patients had heterozygous mutations which correlate with their relatively good acuity and lack of other signs and symptoms. Interestingly both GCD1 and GCD2 can show a phenomenon referred to as granular drop out. This phenomenon can occur after a RCE and subsequent corneal healing resulting in stromal clearing. However, new deposits can then fill in this gap over time.1
GCD2, originally called Avellino corneal dystrophy based on a region of Italy from where two affected families originated, is also an autosomal dominant corneal dystrophy.8 The condition also occurs due to a mutation in the TGFBI gene; however, the mutation in this case is isolated to pArg124His.8,13 This mutation causes the condition initially to have a clinical presentation like GCD1: grey, white granules in the anterior stroma. However, fewer deposits are seen when contrasted with GCD1. The condition tends to be present in early adulthood compared to the first decade of life as seen in GCD1 patients. As GCD2 progresses, the condition tends to differentiate itself from GCD1 as lattice-like amyloid lesions develop.8 These lesions tend to be deeper in the corneal stroma when compared to the granular lesions.7 The lattice presentation is likely owing to a similar genetic etiology for lattice corneal dystrophy and GCD2, mutations in TGFBI. Progression of GCD2 also leads to reduced vision, increased glare and RCE’s like GCD1, although the frequency of RCE is much less compared to RCE frequency in GCD1.14 Similar to GCD1, homozygote patients tend to have a more severe presentation and rapid progression when compared to that experienced by heterozygotes.7
In terms of epidemiology, GCD1 and GCD2 are both rarely encountered; therefore, accurate statistics are not available for the prevalence of the conditions worldwide. However, observations have been made in certain populations. GCD2 has been observed more commonly in Korea and Japan.12,13,15 Specifically, PArg124HIs mutations, which result in GCD2, according to one study were frequently observed in the Japanese population and were responsible for 73% of all corneal dystrophies.16 The second most observed dystrophy in this same population was lattice corneal dystrophy. One large study of affected patients in South Korea, estimated the prevalence of GCD2 to be 11.5 patients per 10,000.13 However, a more recent study estimates the prevalence to be 2 to 3 times higher than the previously mentioned study.15 In contrast, GCD1 is more common in Europe, with statistics not being available.12 Regardless, both conditions are still considered extremely rare. One study did note that GCD was more common among women. However, the study did not classify GCD into subtypes.17 Further studies and analysis are needed to see if gender predilections exist.
Although clinical slit lamp examination (biomicroscopy) is often sufficient to make a diagnosis, at times the clinical signs can be subtle, atypical or overlapping with those of other corneal dystrophies. Therefore, ancillary testing can help clinicians diagnose GCD1 and GCD2 with more accuracy and efficiency. Traditionally, corneal dystrophies were studied using histopathological techniques. However, histological evaluation is not clinically practical. Therefore, in vivo imaging with anterior segment OCT and/or confocal microscopy provides clinicians with a quick, non-invasive and easily accessible way of characterizing corneal dystrophies.
Anterior segment OCT scans and images the corneal layers, in a way similar to a histology section, allowing for the localization of the corneal dystrophy deposits.18 Also noteworthy is that OCT can guide physicians in the surgical management of patients with corneal dystrophies, as the surgeons can determine the depth of the deposits, allowing for the selection of advanced treatments, whether superficial laser treatment or surgery.10,19 Typical OCT findings for GCD1 and GCD2 include delineated hyper reflective lesions that deposit in the anterior two thirds of the stroma, as well as the epithelium and Bowman layers to a lesser extent.10,18,20 This pattern was observed for both patients. Rarely, deposits can extend into the posterior stroma.21 Similar to OCT, confocal microscopy allows for in vivo analysis of the cornea, with images that are comparable to ex vivo histology section.22 Results of confocal imaging in GCD1 and GCD2 are similar to AS OCT, revealing irregular and highly reflective deposits, with clear cornea between them, isolated to the epithelium, Bowmans, and mainly the anterior stroma.21,22 Unfortunately, we did not have access to this technology; however, OCT and genetic testing were adequate to make a diagnosis.
Although the above imaging techniques can help clinicians arrive at a diagnosis, definitive diagnosis can be made with genetic testing, especially in atypical cases. With the introduction of commercially available genetic testing, clinicians now have a method of confirming suspected corneal dystrophies without solely relying on clinical findings, histopathological sections and/or multimodal imaging. Genetic testing is simple to perform as tests today only require a buccal or saliva sample before being sent for analysis. Testing can confirm a tentative diagnosis while also providing information to clinicians and patients on the genetics and inheritance patterns of a suspected condition.23 Ideally, when a corneal dystrophy is confirmed through genetic testing, genetic counselling should be provided to the patient and their family members.24,25 Unfortunately, genetic testing for corneal conditions can be cost prohibitive, as compared to genetic testing for retinal conditions for which testing can be free of charge.26
Differentials
The main differential for GCD1 is GCD2 and vice versa. The types can be differentiated based on their clinical appearance and the results of genetic testing, as mentioned earlier. However other epithelial-stromal corneal dystrophies can present with similar clinical signs and symptoms due to similar mutations in the TGFBI gene.7,8,14
- Lattice corneal dystrophy (LCD): An epithelial stromal corneal dystrophy like GCD1 and GCD2, lattice corneal dystrophy presents with linear and lattice-like amyloid deposits. LCD is also inherited autosomal dominantly and is also due to mutations in the TGFBI gene. However, the mutations in the gene are different when compared to GCD1 and GCD2. In LCD the mutation is in 5q31 locus of the TGFHBI gene, specifically at Arg124Cys.7 Two types of LCD (with various subtypes, respectively) exist. The previously mentioned mutation would be LCD Type 1 while LCD Type 2 is due to mutations in the gelosin gene.7,14 Our patients did not present with lattice or linear corneal signs. Note that the lattice like lines that appear with GCD2 are more dash like than linear, whiter in color than refractile and rarely cross each other when compared to LCD.8 Genetic testing also confirmed the lack of the mutation; therefore, LCD 1 and 2 were ruled out.
- Thiel-Behnke corneal dystrophy (TBCD): Also referred to as honeycomb corneal dystrophy, this condition is linked to Arg555Gln mutations in the TGFBI gene resulting in irregular-shaped deposits that eventually evolve into a honeycomb like appearance.7,14 This pattern was not evident with our two patients and genetic testing excluded this condition.
- Reis Buckler corneal dystrophy: Mutations in the TGFB1 gene at Arg124Leu, which are inherited in an autosomal dominant pattern, result in irregular geographic like deposits that early on can resemble TBCD. A honeycomb or reticular pattern was not seen in our patients with genetic testing also excluding this condition.19,27
Non TGFBI dystrophies can also present similar to GCD1 and GCD2. Although genetic testing is definitive in excluding them, exclusion can also be accomplished with a detailed clinical examination.
- Schnyder’s corneal dystrophy (SCD): SCD is an autosomal dominant stromal corneal dystrophy. However, SCD is not considered a TGFBI corneal dystrophy because its mutation occurs in the UBAID1 gene. This mutation results in the buildup of and deposition of cholesterols and phospholipids in the corneal stroma that can resemble granular deposits. These patients may also present with corneal arcus at an early age owing to elevated serum levels of cholesterol. The cholesterol elevation is also due to UBAID1 gene mutations, as the gene is responsible for cholesterol transport and metabolism. Our patients did not have any cholesterol issues or corneal arcus. Genetic testing also ruled out SCD for both patients.28-30
- Macular corneal dystrophy (MCD): an autosomal recessive stromal dystrophy that manifests secondary to mutations in the CHST6 gene. These mutations results in gray-white central stromal opacities (glycosaminoglycan), that resemble deposits seen in GCD 1 and 2.1,14 With multiple family members in different generations being involved in our cases, this diagnosis was less likely in our patients. Genetic testing ruled out MCD for both patients.
Treatment
Non-Surgical
Glare and reduced visual acuities can result in impairment of activities of daily living for patients if severe enough. Clinicians should consider appropriate correction of refractive error, photochromatic or polarized lenses to minimize glare and a possible referral to low vision providers if surgical intervention is not indicated or preferred by patients.
Non-surgical treatment options also are available, mainly to treat RCE caused by GCD1 and GCD2. The mechanism by which RCE’s occur relates to the position of the granular deposits. Stromal deposits disrupt the hemidesmosomes in corneal basal epithelial cells. These hemidesmosomes are part of an extracellular adhesion complex (along with collagens, metalloproteinases and laminins) that secures the corneal epithelium to the underlying stroma. When this complex is disrupted, the epithelium has a weaker attachment to its basement membrane and is at risk of sloughing off or eroding.31 The epithelium is most vulnerable upon awakening due to the strong adhesive force between the eyelid and cornea from overnight corneal desiccation.11
Treatment of RCE is aimed at reducing friction caused by blinking, promoting epithelial attachment/adhesion, preventing infection and reducing pain. First line therapy for an acute episode is conservative in nature and consists of frequent lubrication using preservative-free artificial tears, nighttime lubricating ointment and antibiotic drops.11 If a patient is in significant pain, a topical cycloplegic drop or bandage contact lens (BCL) can be used. BCL’s offer a twofold benefit in the treatment of RCE: BCLs improve the patient’s symptoms and protect the delicate epithelium and its basement membrane adhesion complex from the overlying eyelid.11,31,32 A prophylactic antibiotic must be used in conjunction with a BCL to prevent secondary infection. Amniotic membranes offer similar advantages to a BCL and can be considered as an alternative, though their cost is significantly greater.11
Once an epithelial erosion has healed, prophylactic treatment to prevent recurrence should be considered. Topical hypertonic sodium chloride ointments or drops promote adhesion of the epithelium and may decrease recurrence by decreasing corneal edema in the intracellular space. Low dose oral doxycycline and topical corticosteroids both prevent breakdown of the corneal extracellular adhesion complex and, therefore, provide stability to the epithelium. Both should be considered in patients unresponsive to first line therapy and/or to prevent recurrence after an acute erosion.33 If an erosion does not heal with traditional therapy, or if recurrent erosions develop within the visual axis, phototherapeutic keratectomy (PTK) can be used and is discussed in more detail below.
Surgical
Corneal laser or surgery are both treatment options when visual acuity is significantly affected or with recurrent corneal erosions that are affecting quality of life in patients with GCD.34 PTK is the least invasive treatment option and can be used when granular deposits are anterior in the stroma. Ablation depth varies with PTK but ranges from 50 to 200 µm, with incomplete deposition clearance occurring more frequently with more shallow ablation depths.27 Hyperopic refractive error, corneal haze and irregular astigmatism due to epithelial hyperplasia are potential complications of PTK.27,35 PTK can be safely repeated and re-treatment is often necessary since recurrence of GCD after PTK is common.35
When PTK is unsuccessful or GCD deposits lie deeper than the anterior stroma, more invasive surgical options are chosen. Anterior lamellar keratoplasty (ALK), deep anterior lamellar keratoplasty (DALK), and penetrating keratoplasty (PK) are all viable treatment options to improve vision.27 ALK can be used to remove mid-stromal deposits. Even deeper deposits, or deposits that form at the graft-host interface after ALK, can be treated with DALK or PK.27,35
Visual recovery time after PTK is quicker than for both lamellar keratoplasties and PK, given its less invasive protocol. In a large, single center study comparing treatment outcomes between PTK, ALK, DALK and PK, patients undergoing PTK achieved their best corrected visual acuity significantly faster, just 1.8 months after the procedure, compared to 5.3 months after PK and 8.4 months after DALK. All patients, no matter the surgical intervention, achieved postoperative final visual acuities between 20/25 and 20/30. These results demonstrate that all surgical options provide favorable outcomes, but visual recovery is quickest with PTK.35 Other published literature supports this finding, with many patients experiencing visual recovery within several weeks after PTK.27,34,36
Recurrence of GCD is well-reported and can occur after any surgical intervention. Recurrence occurs centrally and anteriorly in the cornea.35 While PTK offers quick visual recovery, patients experience recurrence sooner than with deeper surgical interventions.35 In the previously mentioned study by Lewis et al, the median length of time until significant recurrence after PTK was 2.7 years, as compared to the median time to significant recurrence after PK of 13.7 years. Both lamellar keratoplasties had similar lengths until significant recurrence at 3.7 years (ALK) and 3.2 years (DALK).35 Genetic status has been proposed as playing a role in rates of recurrence, with homozygous patients experiencing recurrence sooner than heterozygotes.27,35 This proposal aligns with the previously described clinical course of homozygotes having a more severe clinical presentation. Note that PTK and lamellar keratoplasty (LK) can both be performed safely on graft tissue after a PK, if recurrence is noted.34,37,38 LK is often preferred to PTK in these instances, as consecutive PTK’s flatten and thin the graft tissue, leading to a higher risk of hyperopia and ectasia development.38 Furthermore, data suggests that subsequent PTK retreatments on PK grafts are performed at shorter intervals, indicating that recurrence may occur more rapidly after each consecutive treatment.37
Penetrating keratoplasty (PK) was the only form of surgical treatment for many years and may offer a longer interval until recurrence, but PK is a more invasive procedure compared to LK and PTK, requiring full host tissue removal. Lamellar keratoplasty is less invasive and offers an advantage over PTK in removing deeper deposits, but patients may still require repeat procedures and take longer to visually recover compared to treatment with PTK alone.35 Selection of a surgical treatment must weigh the rate of visual recovery against the potential for recurrence and need for retreatment. The genetic composition of a patient’s GCD and the depth of their granular deposits also play a role in surgical selection.
Genetic Treatment Options
Because the cornea is easily accessed and imaged, the tissue is a viable target of genetic research. In addition, the cornea is immune-privileged which allows for easy gene transfer.23 However, as promising as gene therapy is, especially for retinal conditions in which surgery is not option, surgical success for corneal conditions is likely limiting the introduction and production of possible gene therapies for corneal dystrophies. Surgical options in the past have included the excising and replacement of the entire cornea (PK). With advents in technology, specific layers of the cornea can be removed, with the potential for good visual outcomes within weeks of surgery.27 However, granular deposits may recur as mentioned earlier; thus, surgery exists as a treatment option and not a cure.8,39 Further studies are needed at this time. Therefore, the viability of future genetic treatment options, including gene editing with newer technologies, such as clustered regularly interspaced short palindromic repeats (CRISPR), will be vital going forward.40
Conclusion
GCD 1 and GCD2 are rare autosomal dominantly inherited corneal epithelial stromal dystrophies. Early in the disease process the conditions may appear similar; therefore, clinicians should be cognizant of differentiating the individual disease processes as the two conditions progress. In addition, many other corneal dystrophies can mimic GCD 1 and/or 2. In vivo imaging can aid clinicians in arriving at a faster diagnosis, and genetic testing is the ultimate confirmatory tool. Although no cure exists, sequala such as reduced visual acuity and corneal erosions can be managed through non-surgical and surgical means.
References
- Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies–edition 2. Cornea. 2015 Feb;34(2):117-59. DOI: 10.1097/ico.0000000000000307
- Bhaduri G. Gyrate atrophy of choroid and retina. J Indian Med Assoc. 2002 Mar;100(3):196-7.
- Lyons CJ, McCartney AC, Kirkness CM, Ficker, LA, Steele, AD, Rice, NS. Granular Corneal Dystrophy: Visual Results and Pattern of Recurrence after Lamellar or Penetrating Keratoplasty. Ophthalmology. 1994 Nov;101(11):1812-7. DOI: 10.1016/S0161-6420(94)31096-7
- Roncone DP. Granular corneal dystrophy: a novel approach to classification and treatment. Optom Vis Sci. 2014 Mar;91(3):e63-71. DOI:10.1097/opx.0000000000000159
- Wilson CM, D’Ath PJ, Parmar DN, Sykakis E. Keratoconus and granular dystrophy. BMJ Case Rep. 2014 Aug 25;2014DOI:10.1136/bcr-2014-205584
- Weiss JS, Møller HU, Lisch W, et al. The IC3D classification of the corneal dystrophies. Cornea. 2008 Dec;27 Suppl 2(Suppl 2):S1-83. DOI:10.1097/ICO.0b013e31817780fb
- Han KE, Choi SI, Kim TI, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Prog Retin Eye Res. 2016 Jan;50:67-88. DOI:10.1016/j.preteyeres.2015.11.002
- Chang MS, Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean J Ophthalmol. 2023 Aug;37(4):340-7. DOI:10.3341/kjo.2023.0032
- Weidle EG, Lisch W. Die verschiedenen Trübungsformen der bröckeligen Hornhautdystrophie. [Various forms of opacities of granular corneal dystrophy]. Klin Monbl Augenheilkd. 1984 Sep;185(3):167-73. DOI:10.1055/s-2008-1054592
- Miura M, Mori H, Watanabe Y, et al. Three-Dimensional Optical Coherence Tomography of Granular Corneal Dystrophy. Cornea. 2007 Apr;26(3) DOI: 10.1097/ICO.0b013e31802e1e50
- Miller DD, Hasan SA, Simmons NL, Stewart MW. Recurrent corneal erosion: a comprehensive review. Clin Ophthalmol. 2019 Feb 11;13:325-35. DOI:10.2147/opth.s157430
- Klintworth GK. Corneal dystrophies. Orphanet J Rare Dis. 2009 Feb 23;4:7. DOI:10.1186/1750-1172-4-7
- Lee JH, Cristol SM, Kim WC, et al. Prevalence of granular corneal dystrophy type 2 (Avellino corneal dystrophy) in the Korean population. Ophthalmic Epidemiol. 2010 Jun;17(3):160-5. DOI:10.3109/09286581003624939
- Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Exp Eye Res. 2019 Sep;186:107715. DOI:10.1016/j.exer.2019.107715
- Park JE, Yun SA, Roh EY, Yoon JH, Shin S, Ki,CS. Prevalence of granular corneal dystrophy type 2-related TGFBI p.R124H variant in a South Korean population. Mol Vis. 2021 May 8;27:283-7.
- Fujiki K, Nakayasu K, Kanai A. Corneal dystrophies in Japan. J Hum Genet. 2001;46(8):431-435. DOI:10.1007/s100380170041
- Musch DC, Niziol LM, Stein JD, Kamyar RM, Sugar A. Prevalence of corneal dystrophies in the United States: estimates from claims data. Invest Ophthalmol Vis Sci. 2011 Sep 1;52(9):6959-63. DOI:10.1167/iovs.11-7771
- Dalton K, Schneider S, Sorbara L, Jones L. Confocal microscopy and optical coherence tomography imaging of hereditary granular dystrophy. Contact Lens and Anterior Eye. 2010/02/01/ 2010;33(1):33-40. DOI: 10.1016/j.clae.2009.09.005
- Elhardt C, Priglinger SG, Karakolova Y, Mayer WJ, Wertheimer C. Hornhautdystrophien in der optischen Kohärenztomographie. [Corneal dystrophies in optical coherence tomography]. Ophthalmologe. 2019 Sep;116(9):857-64. DOI: 10.1007/s00347-018-0832-8
- Siebelmann S, Scholz P, Sonnenschein S, et al. Anterior segment optical coherence tomography for the diagnosis of corneal dystrophies according to the IC3D classification. Survey of Ophthalmology. 2018 May-Jun;63(3):365-80. DOI: 10.1016/j.survophthal.2017.08.001
- Cortes D, Li J, Chen M, et al. High-Resolution Anterior Segment Optical Coherence Tomography and in vivo Confocal Microscopy in the Evaluation of Corneal Dystrophies. Invest Ophthalmol Vis Sci. 2013;54(15):562.
- Shukla AN, Cruzat A, Hamrah P. Confocal microscopy of corneal dystrophies. Semin Ophthalmol. 2012 Sep-Nov;27(5-6):107-16. DOI:10.3109/08820538.2012.707276
- Weiss JS, Willoughby CE, Abad–Morales V, Turunen JA, Lisch W. Update on the Corneal Dystrophies—Genetic Testing and Therapy. Cornea. 2022 Nov 1;41(11):1337-1344. DOI: 10.1097/ICO.0000000000002857
- Hui EKY, Yam JCS, Rahman F, Pang CP, Kumaramanickavel G. Ophthalmic genetic counselling: emerging trends in practice perspectives in Asia. J Community Genet. 2023 Feb;14(1):81-9. DOI:10.1007/s12687-022-00616-w
- Alabek M, Andersen K, Everett L, Marra M. The genetic counselor workforce in inherited retinal disease clinics: a descriptive assessment. Ophthalmic Genet. 2023 Dec;44(6):553-8. DOI:10.1080/13816810.2023.2239910
- McClard CK, Pollalis D, Jamshidi F, Kingsley R, Lee SY. Utility of No-Charge Panel Genetic Testing for Inherited Retinal Diseases in a Real-World Clinical Setting. J Vitreoretin Dis. 2022 Sep-Oct;6(5):351-7. DOI:10.1177/24741264221100936
- Ashena Z, Niestrata M, Tavassoli S. Management of Stromal Corneal Dystrophies; Review of the Literature with a Focus on Phototherapeutic Keratectomy and Keratoplasty. Vision. 2023 Mar 13;7(1). DOI:10.3390/vision7010022
- Evans CJ, Dudakova L, Skalicka P, et al. Schnyder corneal dystrophy and associated phenotypes caused by novel and recurrent mutations in the UBIAD1 gene. BMC Ophthalmology. 2018 Sep 17;18(1):250. DOI:10.1186/s12886-018-0918-8
- Ghazal W, Georgeon C, Grieve K, Bouheraoua N, Borderie V. Multimodal Imaging Features of Schnyder Corneal Dystrophy. J Ophthalmol. 2020 Mar 23;2020:6701816. DOI:10.1155/2020/6701816
- Weiss JS. Schnyder corneal dystrophy. Curr Opin Ophthalmol. 2009 Jul;20(4):292-8. DOI:10.1097/ICU.0b013e32832b753e
- Lin SR, Aldave AJ, Chodosh J. Recurrent corneal erosion syndrome. Br J Ophthalmol. 2019 Sep;103(9):1204-1208. DOI:10.1136/bjophthalmol-2019-313835
- Nanba H, Mimura T, Mizuno Y, et al. Clinical course and risk factors of recurrent corneal erosion: Observational study. Medicine (Baltimore). 2019 Apr;98(16):e14964. DOI:10.1097/md.0000000000014964
- Ramamurthi S, Rahman MQ, Dutton GN, Ramaesh K. Pathogenesis, clinical features and management of recurrent corneal erosions. Eye (Lond). 2006 Jun;20(6):635-44. DOI:10.1038/sj.eye.6702005
- Camesasca FI, Vinciguerra R, Legrottaglie EF, Morenghi E, Trazza S, Vinciguerra P. Sequential Custom Therapeutic Keratectomy for the Treatment of Granular Corneal Dystrophy Type 1: A Long-term Study. J Refract Surg. 2023 Jun;39(6):422-9. DOI:10.3928/1081597x-20230503-01
- Lewis DR, Price MO, Feng MT, Price FW Jr. Recurrence of Granular Corneal Dystrophy Type 1 After Phototherapeutic Keratectomy, Lamellar Keratoplasty, and Penetrating Keratoplasty in a Single Population. Cornea. 2017 Oct;36(10):1227-32. DOI:10.1097/ico.0000000000001303
- Reddy JC, Rapuano CJ, Nagra PK, Hammersmith KM. Excimer laser phototherapeutic keratectomy in eyes with corneal stromal dystrophies with and without a corneal graft. Am J Ophthalmol. 2013 Jun;155(6):1111-8.e2. DOI:10.1016/j.ajo.2012.12.016
- Rathi VM, Taneja M, Murthy SI, Bagga B, Vaddavalli PK, Sangwan VS. Phototherapeutic keratectomy for recurrent granular dystrophy in postpenetrating keratoplasty eyes. Indian J Ophthalmol. 2016 Feb;64(2):140-4. DOI:10.4103/0301-4738.179715
- Taneja M, Rathi VM, Murthy SI, Bagga B, Vaddavalli PK. Femtosecond Laser-Assisted Anterior Lamellar Keratoplasty for Recurrence of Granular Corneal Dystrophy in Postkeratoplasty Eyes. Cornea. 2017 Mar;36(3):300-3. DOI:10.1097/ico.0000000000001068
- Sahay P, Agarwal D, Maharana PK, Titiyal JS. Granular corneal dystrophy: an enigma resolved. Int Ophthalmol. 2019 Jul;39(7):1599-1602. DOI:10.1007/s10792-018-0971-6
- Mohan RR, Martin LM, Sinha NR. Novel insights into gene therapy in the cornea. Exp Eye Res. Jan 2021;202:108361. DOI: 10.1016/j.exer.2020.108361

